Open Materials 2024 - A Foundation Model for Inorganic Materials Modeling
Update: following Meta FAIR's release of Open Molecules 2025 (OMol25), we have deprecated OMat24 eqV2. OMol25 is a high-quality DFT dataset of unprecedented scale spanning small molecules, biomolecules, metal complexes, and electrolytes, including 83 elements, charged systems, and open-shell species. Run models trained on OMol25 on Rowan today.
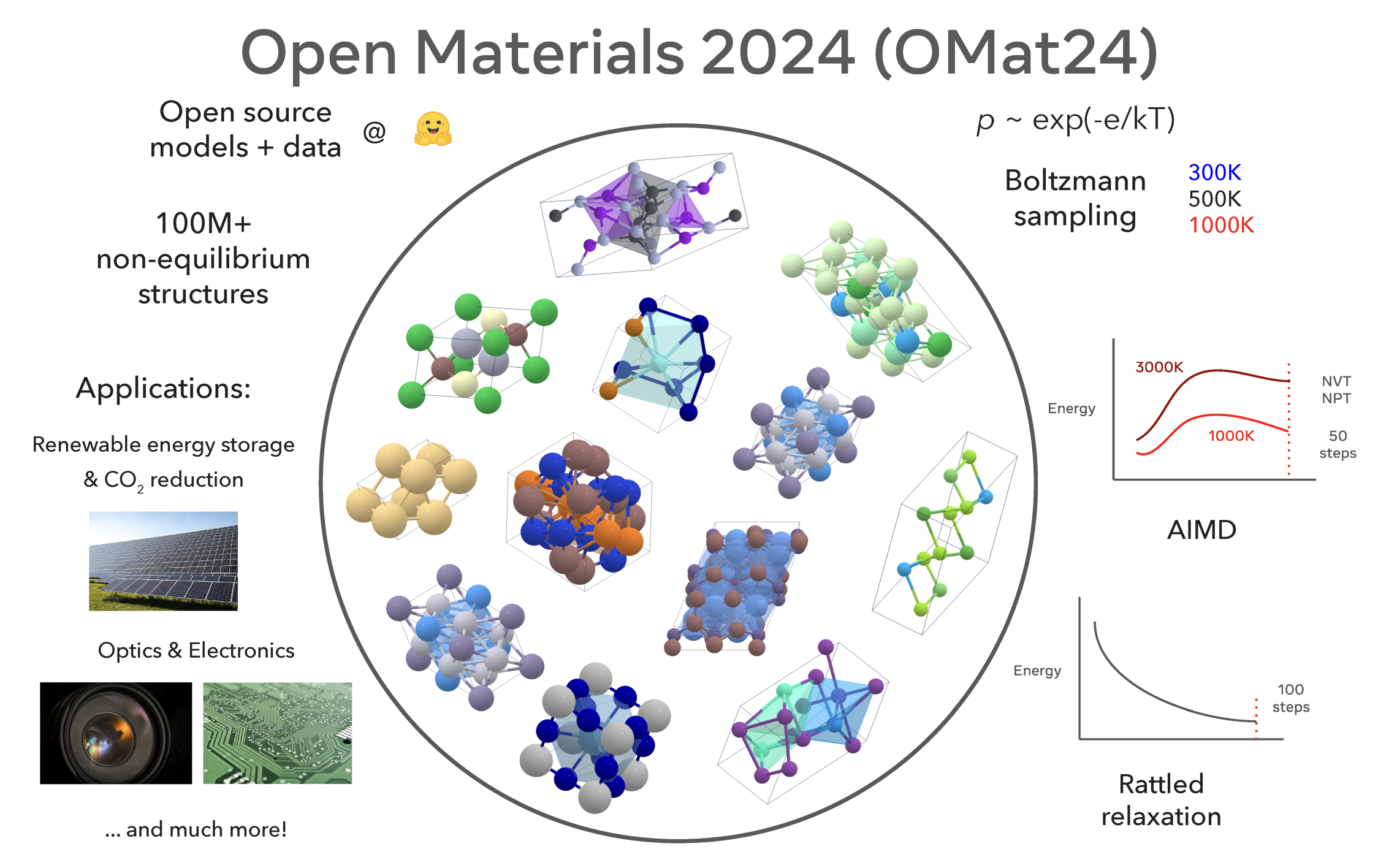
Open Materials 2024, henceforth abbreviated OMat24, is a massive DFT dataset and a machine learning-based interatomic potential trained to reproduce the results of this dataset. OMat24 is the latest from the FAIR-chem group at Meta, and was released in October 2024. Here's an overview of the dataset, the model, and why we're so excited to bring this to our users.
Decent Low-Cost Models Of Materials Haven't Existed
Unlike models focused on organic chemistry or solution-phase molecular interactions, like AIMNet2, BAMBOO, MACE-OFF23, and plenty of others, the OMat24 models are specifically focused on reproducing the results of periodic DFT calculations for bulk materials. This has historically been very difficult for low-cost methods: the forcefields commonly used to model bioorganic systems don't extend to these systems, and what forcefields do exist are very inaccurate (like UFF). Semiempirical methods like xTB have emerged as an alternative in recent years, but support for periodic systems is irregular and convergence can be challenging.
If you want to run reasonably accurate simulations of materials, then, your only option has been periodic DFT, which is infamously compute-intensive. OMat24 changes this: it has reasonable accuracy, broad applicability, and runs orders of magnitude faster than the corresponding DFT calculations would, allowing meaningful simulation throughput to be achievable on small computers.
The OMat24 Dataset
In machine learning, "the dataset is the model"—good datasets allow modern architectures to shine, while bad datasets lead to unreliable results and poor performance in the real world. While datasets have been getting larger in computational chemistry, they still fall far short of the massive datasets that have been necessary for breakthroughs in language modeling or computer vision.
OMat24 changes this. The authors ran over 100 million periodic DFT calculations on a wide variety of compositions and structures, which is about two orders of magnitude larger than the previously state-of-the-art MPtrj dataset. And these aren't just more of the same type of calculation: OMat24 used a variety of techniques to sample non-equilibrium structures, including rattled Boltzmann sampling, ab initio molecular dynamics, and rattled relaxation. As a result, the dataset has many more structures with non-zero forces and stress than previous datasets, which should lead to better performance in molecule dynamics and higher robustness overall.
The dataset isn't perfect, of course. OMat24 uses the PBE functional (with optional Hubbard U correction), which is standard in materials modeling but which has a number of known pathologies—it's quite bad for organic thermochemistry, for instance, and the description of bulk water is pretty poor. As such, OMat24 won't be the last dataset needed for training models in computational chemistry. But it's still a huge leap forward for the field, and will doubtless prove to be incredibly impactful in the years to come.
The OMat24 Models
The authors train a variety of neural network potentials on this dataset. The potentials are based on the EquiformerV2 architecture, which is a message-passing equivariant graph neural network developed by Yi-Lun Liao and Tess Smidt at MIT, and use the new non-equilibrium denoising technique to improve data efficiency. The scale and diversity of the dataset allows the authors to train NNPs of unprecedented size—their largest model has around 150 million parameters, which is significantly larger than previous pretrained models for materials modeling, like MACE-MP-0.
The resulting models are best-in-class for almost every metric studied, as surveyed by the Matbench Discovery leaderboard. For instance, OMat24 increases the positive rate of identifying thermodynamic stability to above 90% for the first time, and achieves an F1 score of 0.917 (versus a previous best of 0.880). The authors note that OMat24 closely approaches the accuracy of the underlying PBE theory in many cases, and argue that higher accuracy training data is needed for further progress:
...discrepancies between the PBE functional and experimental results limit the effective value of predictions with respect to experiments. Developing training sets and benchmarks using more accurate DFT functionals such as SCAN and r2SCAN could help overcome these challenges.
The OMat24 models not only replicate the results of existing DFT on materials, but do so while running many orders of magnitude faster and exhibiting better simulation stability. No tricky SCF convergence is observed with OMat24, for instance, so force predictions can be generated in a single forward inference step no matter the system.
When and How To Use OMat24
It's important to note that OMat24 isn't a full replacement for DFT yet: the scope of systems that it seeks to model is massive, and lots more data (and possibly new architectures) will be needed to achieve great performance across all possible materials. There's other limiting factors here, too: the PBE functional that predominates in materials science is quite inaccurate for barrier heights and many intermolecular interactions, so some form of fancier multi-fidelity learning will probably be necessary to achieve high accuracy in the long run.
But even so, OMat24 is a huge leap forward for the field: it's fast, reliable, and gives extremely useful results in a tiny fraction of the time that competing methods would take. If you're looking to simulate a problem in materials science, there's no reason not to try OMat24 out and see how it performs!
If you want to try running a calculation with OMat24 right now, create an account on Rowan and start within minutes!
