Run molecular design, simulation, and prediction workflows through a scientist-friendly web app or a structured API. Rowan helps teams in drug discovery and materials science move faster without building infrastructure from scratch.
Get the capabilities of a modern scientific platform without the infrastructure overhead.
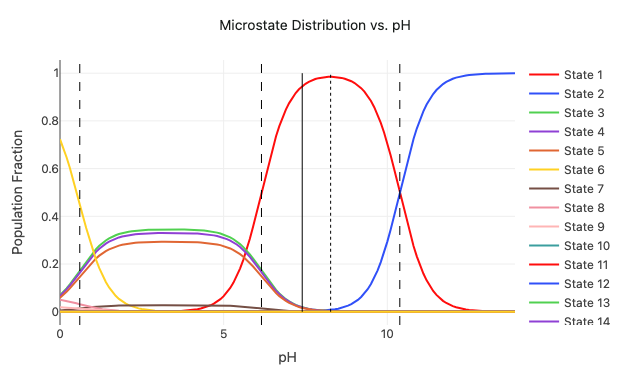
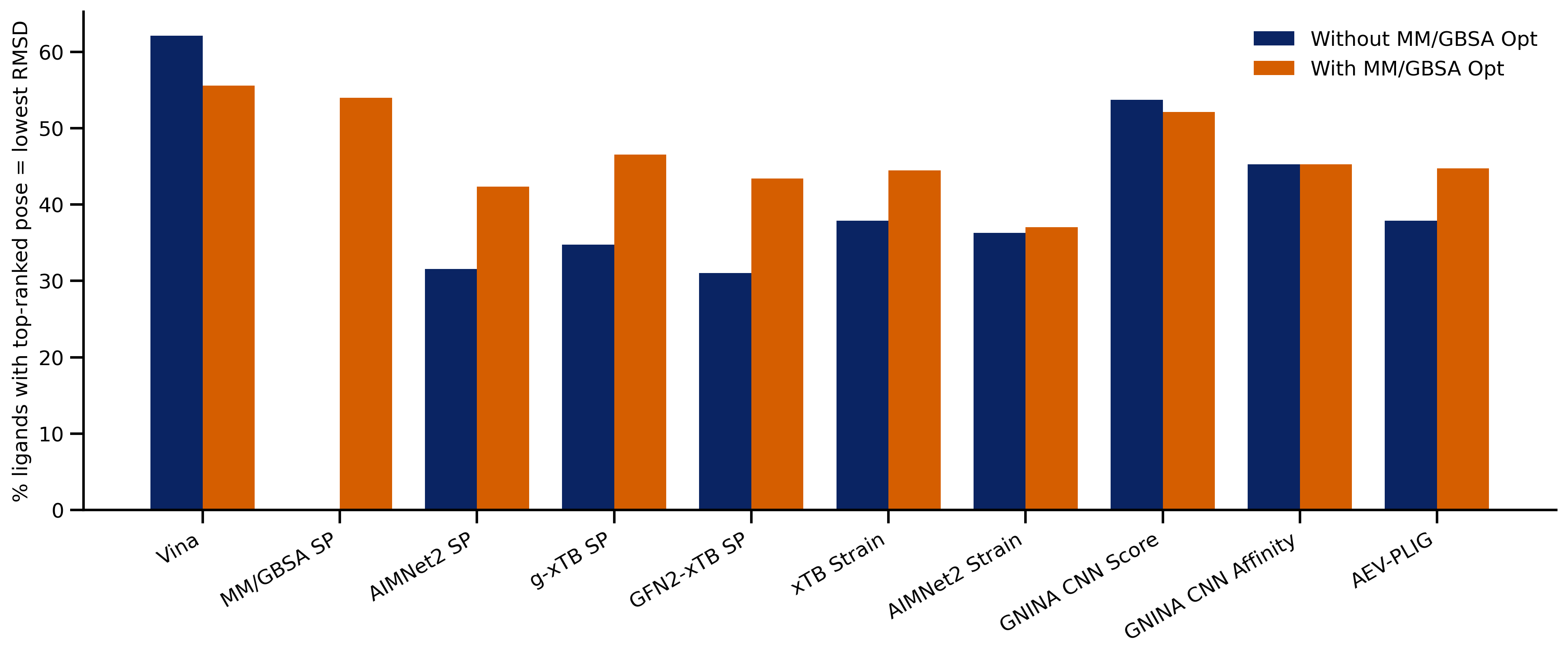
Predict molecular properties with end-to-end workflows to predict acidity (pKa), bond strength (BDE), reduction/oxidation potentials, and more.
Accelerate your molecular simulation with neural network potentials like UMA or use industry-standard methods like xTB and GPU-accelerated DFT.
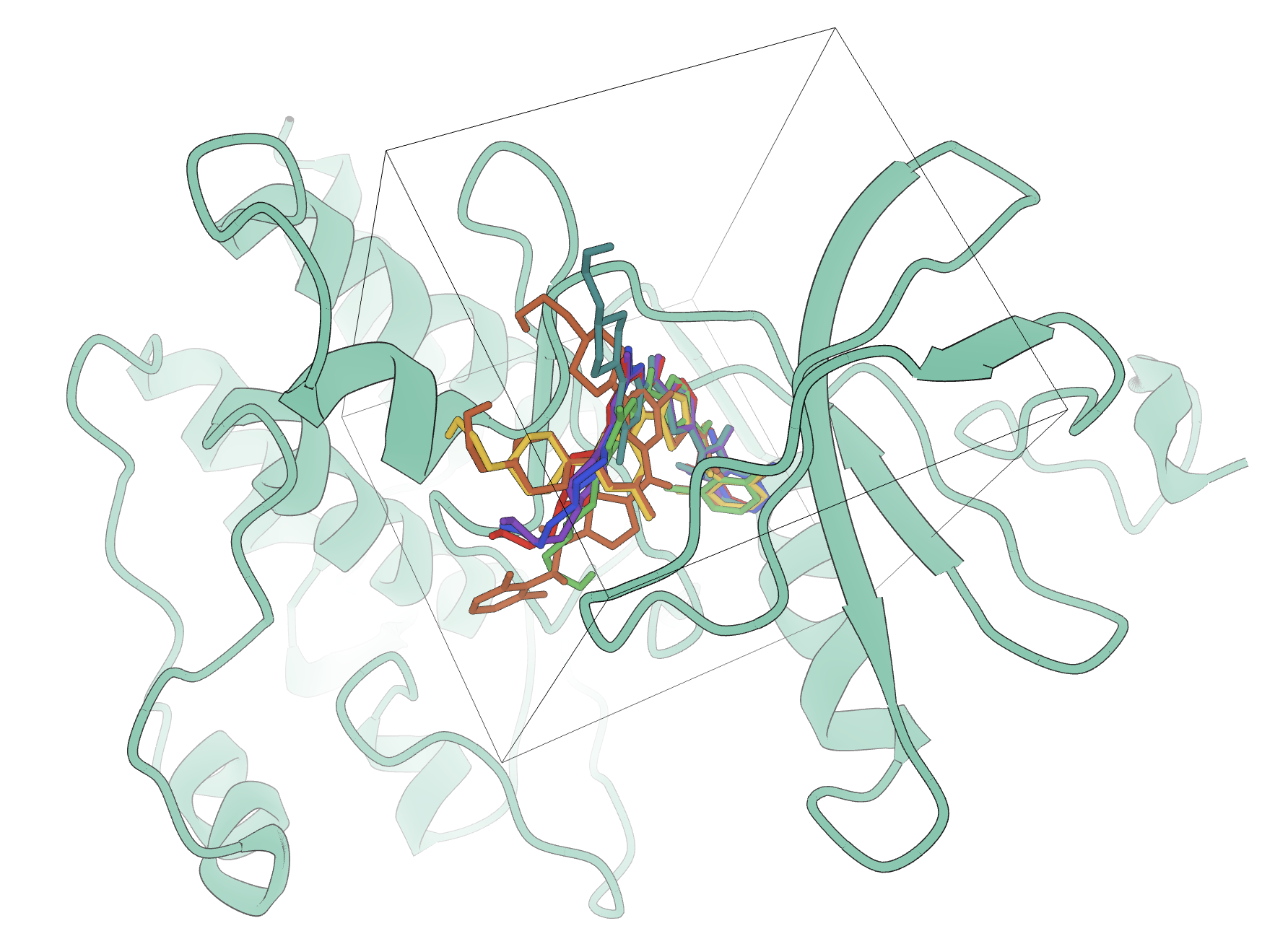
Screen large molecular libraries in silico so experimental work starts from stronger candidates.
Learn more →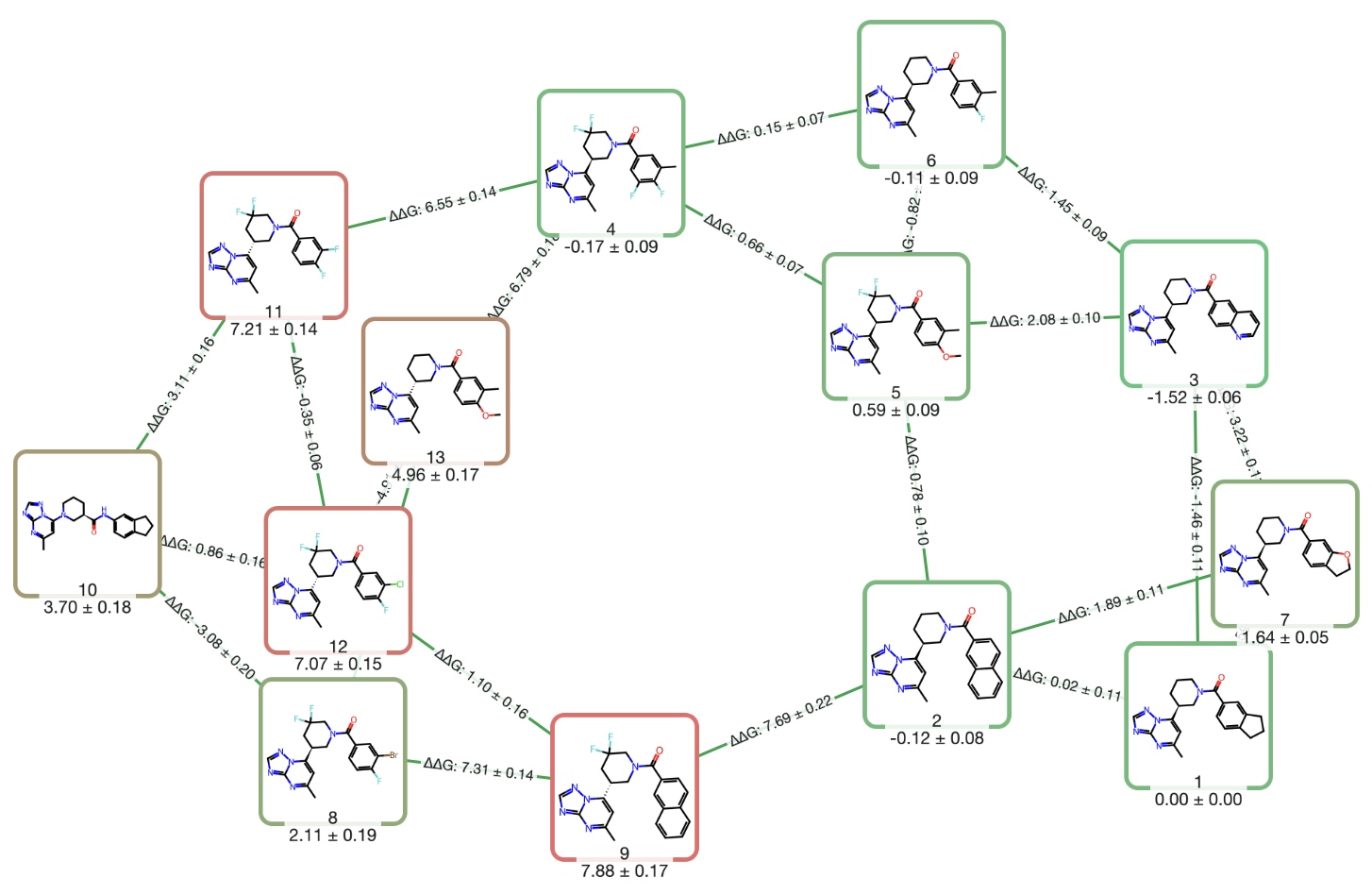
Use RBFE calculations to accurately predict protein–ligand binding affinity and optimize potency & selectivity.
Learn more →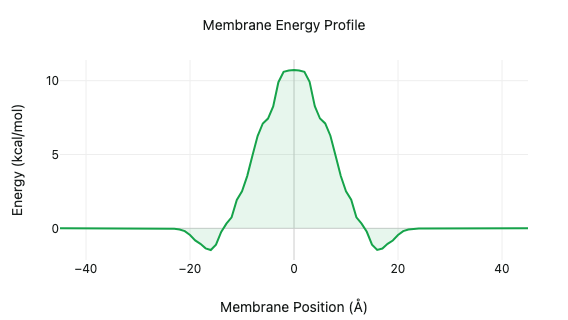
Predict solubility, permeability, and blood–brain barrier penetration with physics-based and machine-learning models.
Investigate reaction mechanisms with automated transition-state search, Fukui indices, and IRC validation.
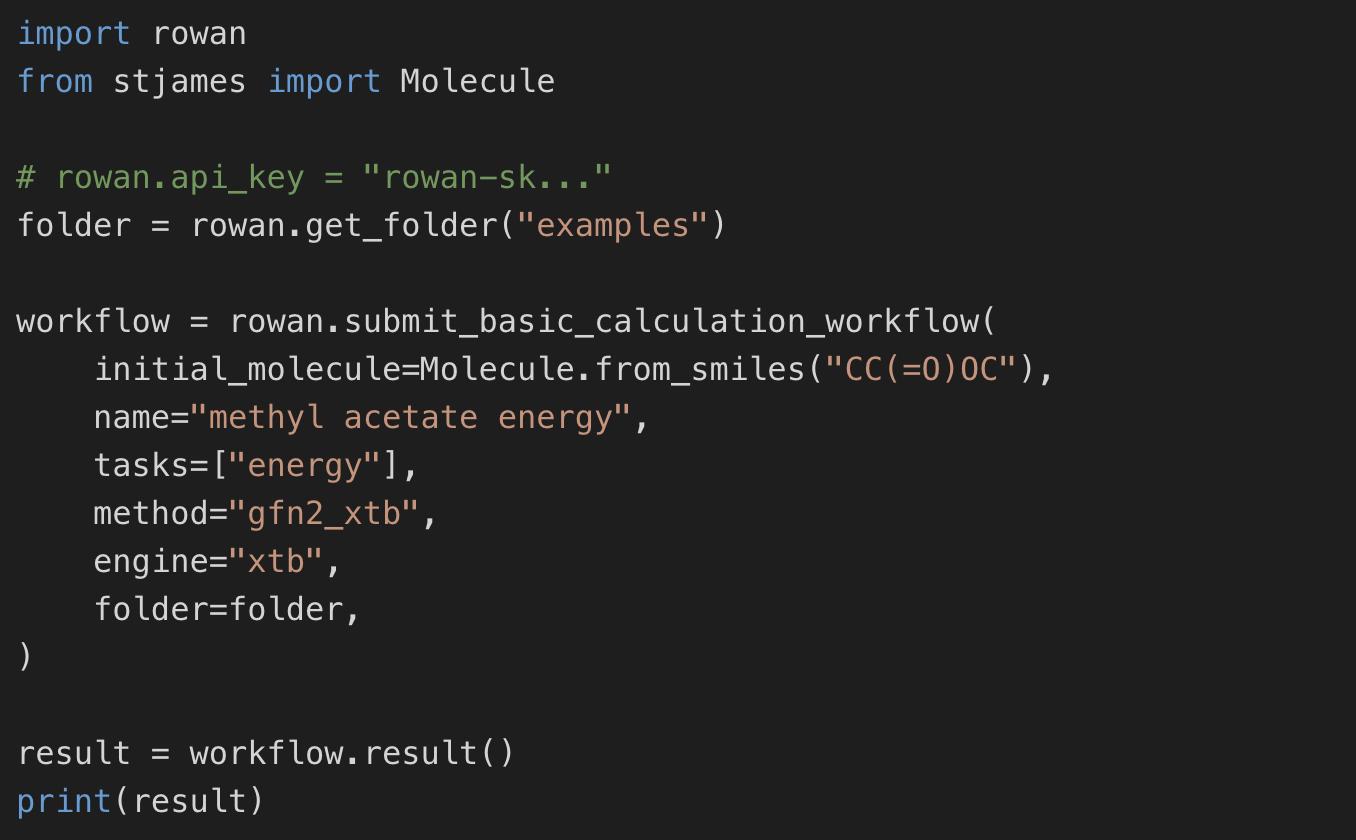
Run Rowan through our Python API or integrate it into automated workflows. Structured results make it easy to screen large libraries and build complex pipelines.
Learn more →Your intellectual property remains yours. Rowan also supports single-tenant and customer-managed deployments for teams with stricter security and infrastructure requirements.
Learn more →Trusted by
14K+14,000+
scientists
Over
2.5M2.5 million
calculations run
Move from large-scale screening to candidate selection in one computational environment.
Hear what scientists have to say.
Rowan makes it easy for us to run reliable computational workflows without building or maintaining our own compute infrastructure. This helps us move faster across Curie's portfolio companies while keeping the focus on advancing the pipeline. The new solvent-dependent conformer workflow is an exciting, impactful addition.
Read the latest posts from our newsletter and blog.

Create an account to get started today or contact us to find a solution for your business.