Efficient and Accurate pKa Prediction Enabled by Pre-Trained Machine-Learned Interatomic Potentials
Corin C. Wagen and Arien M. Wagen
Mar 8, 2024
Quickly and accurately predicting the pKa of small molecules is an important unsolved challenge in computational chemistry: while approaches based on electronic structure theory have shown great promise, the utility of these methods is limited by the considerable expense of the requisite computations. In this study, we investigate AIMNet2, a machine-learned interatomic potential, as a low-cost surrogate for electronic structure theory in pKa prediction. The accuracy of the AIMNet2-based pKa prediction workflow is evaluated over a wide range of compounds and functional groups, and potential sources of error are discussed.
Knowing the pKa of a molecule is key to understanding its structure and reactivity. In medicinal chemistry, pKa values are used to predict a variety of important pharmacological properties, including solubility, hERG potassium channel blocking, phospholipidosis risk, CYP inhibition, and blood–brain-barrier penetration.1–61. Di, L.; Kerns, E. H. Drug-Like Properties: Concepts, Structure Design, and Methods, 2nd ed.; Academic Press, 2016. 2. Charifson, P. S.; Walters, W. P. Acidic and Basic Drugs in Medicinal Chemistry: A Perspective. J. Med. Chem.2014, 57, 9701–9717. 3. Garrido, A. et al. hERG toxicity assessment: Useful guidelines for drug design. Eur. J. Med. Chem.2020, 195, 112290. 4. Ploeman, J.-P. H.T.M. et al. Use of physicochemical calculation of pKa and CLogP to predict phospholipidosis-inducing potential: A case study with structurally related piperazines. Exp. Toxicol. Pathol.2004, 55, 347–355. 5. Pelletier, D. J. et al. Evaluation of a Published in Silico Model and Construction of a Novel Bayesian Model for Predicting Phospholipidosis Inducing Potential. J. Chem. Inf. Model.2007, 47, 1196–1205. 6. Manallack, D. T. The pKa Distribution of Drugs: Application to Drug Discovery. Perspect. Medicin. Chem.2007, 1, 25–38. Efficient and accurate pKa estimates play a pivotal role in the design of compounds with the desired physicochemical and pharmacological properties, and accordingly pKaprediction has been recognized as a problem of "immense interest"77. Meloun, M.; Bordovská, S. Benchmarking and validating algorithms that estimate pKa values of drugs based on their molecular structures. Anal. Bioanal. Chem.2007, 389, 1267–1281. in computational chemistry for decades.8,98. Liao, C.; Nicklaus, M. C. Comparison of Nine Programs Predicting pKa Values of Pharmaceutical Substances. J. Chem. Inf. Model.2009, 49, 2801–2812. 9. Settimo, L.; Bellman, K.; Knegtel, R. M. A. Comparison of the Accuracy of Experimental and Predicted pKa Values of Basic and Acidic Compounds. Pharm. Res.2013, 31, 1082–1095.
The most common approach to pKa prediction is to develop an empirical quantitative structure–property relationship (QSPR) based on experimentally measured data and use this model to predict pKa values for new compounds.1010. For state-of-the-art QSPR methods, see: (a) Johnston, R. C. et al. Epik: pKa and Protonation State Prediction through Machine Learning. ChemRxiv, 2023, 10.26434/chemrxiv-2023-c6z8t-v3. (b) Mayr, F.; Wieder, M.; Wieder, O.; Langer, T. Improving Small Molecule pKa Prediction Using Transfer Learning With Graph Neural Networks. Front. Chem.2022, 10, 866585. (c) Luo, W. et al. Uni-pKa: An Accurate and Physically Consistent pKa Prediction through Protonation Ensemble Modeling. ChemRxiv, 2023, 10.26434/chemrxiv-2023-lw5k0. QSPR approaches are typically fast and display excellent accuracy for compounds like those in the training data, making them an appealing choice for high-throughput pKa prediction.1111. Rupp, M.; Körner, R.; Tetko, I. V. Predicting the pKa of Small Molecules. Comb. Chem. High Throughput Screen.2011, 14, 307–327. However, these methods struggle to extrapolate to unseen compounds and are generally unable to capture factors that arise from specific configurations of molecules in three-dimensional space, like conformational or stereoelectronic effects.1212. Navo, C. D.; Jiménez-Osés, G. Computer Prediction of pKa Values in Small Molecules and Proteins. ACS Med. Chem. Lett.2021, 12, 1624–1628.
A contrasting approach is to directly predict pKa values using electronic structure theory, since microscopic pKa values are directly proportional to the ∆G of the underlying acid dissociation reactions. This strategy possesses certain advantages over QSPR approaches: conformational and stereochemical effects are naturally incorporated, and new regions of chemical space can be modeled without new training data. In recent pKa prediction challenges, approaches based on electronic structure theory have consistently been among the best-performing methods: in SAMPL6, submission xvxzd achieved a mean absolute error (MAE) of 0.58 pKa units and a root-mean-squared error (RMSE) of 0.68,1313. Işık, M. et al. Overview of the SAMPL6 pKa Challenge: Evaluating small molecule microscopic and macroscopic pKa predictions. J. Comput. Aided Mol. Des.2021, 35, 131–166. while submission EC_RISM achieved an MAE of 0.53 and an RMSE of 0.73 in SAMPL7.1414. Bergazin, T. D. et al. Evaluation of log P, pKa, and log D predictions from the SAMPL7 blind challenge. J. Comput. Aided Mol. Des.2021, 35, 771–802.
Unfortunately, many electronic structure theory-based prediction approaches are too computationally costly for routine usage: in their paper describing the xvxzd submission to SAMPL6, Pracht and Grimme write that "a macroscopic pKa value for a molecule could be calculated within approximately one day on a 28 CPU node,"1515. Pracht, P. et al. High accuracy quantum-chemistry-based calculation and blind prediction of macroscopic pKa values in the context of the SAMPL6 challenge. J. Comput. Aided Mol. Des.2018, 32, 1139–1149. which corresponds to roughly $25–30 with current cloud computing prices.1616. Based on current AWS EC2 on-demand pricing (Feb. 26, 2024) a c5.4xlarge instance with 16 vCPUs costs $0.68/hour: $0.68/hour * 28/16 * 24 hours = $28.56. While semiempirical methods like PM61717. Kromann, J. C.; Larsen, F.; Moustafa, H.; Jensen, J. H. Prediction of pKa values using the PM6 semiempirical method. PeerJ, 2016, 4, e2335. and GFN2-xTB1818. Pracht, P.; Grimme, S. Efficient Quantum-Chemical Calculations of Acid Dissociation Constants from Free-Energy Relationships. J. Phys. Chem. A.2021, 125, 5681–5692. have been used for rapid pKaprediction, the resulting workflows display lower accuracy and often require judicious choice of reference compound or significant reparameterization. This is unsurprising: calculation of acid dissociation constants is "demanding and arduous,"1919. Alongi, K. S.; Shields, G. C. Theoretical Calculations of Acid Dissociation Constants: A Review Article. In Annual Reports in Computational Chemistry,; Wheeler, R. A., Ed.; Elsevier, 2010; Vol. 6, 113–138. and both a sophisticated electronic structure method and a sizable basis set are required to consistently get accurate results even in the gas phase.2020. Dékány, A. A.; Czakó, G. Benchmark ab initio proton affinity and gas-phase basicity of α-alanine based on coupled-cluster theory and statistical mechanics. J. Comput. Chem.2021, 43, 19–28.
Machine-learned interatomic potentials have recently emerged as an efficient alternative to conventional electronic structure theory.2121. See, inter alia, (a) Behler, J. Four Generations of High-Dimensional Neural Network Potentials. Chem. Rev.2021, 121, 10037–10072. (b) Friederich, P.; Häse, F.; Proppe, J.; Aspuru-Guzik, A. Machine-learned potentials for next-generation matter simulations. Nat. Mater.2021, 20, 750–761. (c) Anstine, D. M.; Isayev, O. Machine Learning Interatomic Potentials and Long-Range Physics. J. Phys. Chem. A.2023, 127, 2417–2431. We envisioned that AIMNet2, which displays accuracy comparable to routine density-functional theory methods for main-group thermochemistry,2222. Anstine, D. M.; Zubatyuk, R.; Isayev, O. AIMNet2: A Neural Network Potential to Meet your Neutral, Charged, Organic, and Elemental-Organic Needs. ChemRxiv, 2023, 10.26434/chemrxiv-2023-296ch. might allow for rapid computation of pKavalues while preserving the advantages of electronic structure-based approaches. Here, we report the successful implementation of this strategy into a workflow ("Rowan pKa"), which uses AIMNet2 to calculate microscopic pKa values with good accuracy and minimal empiricism.
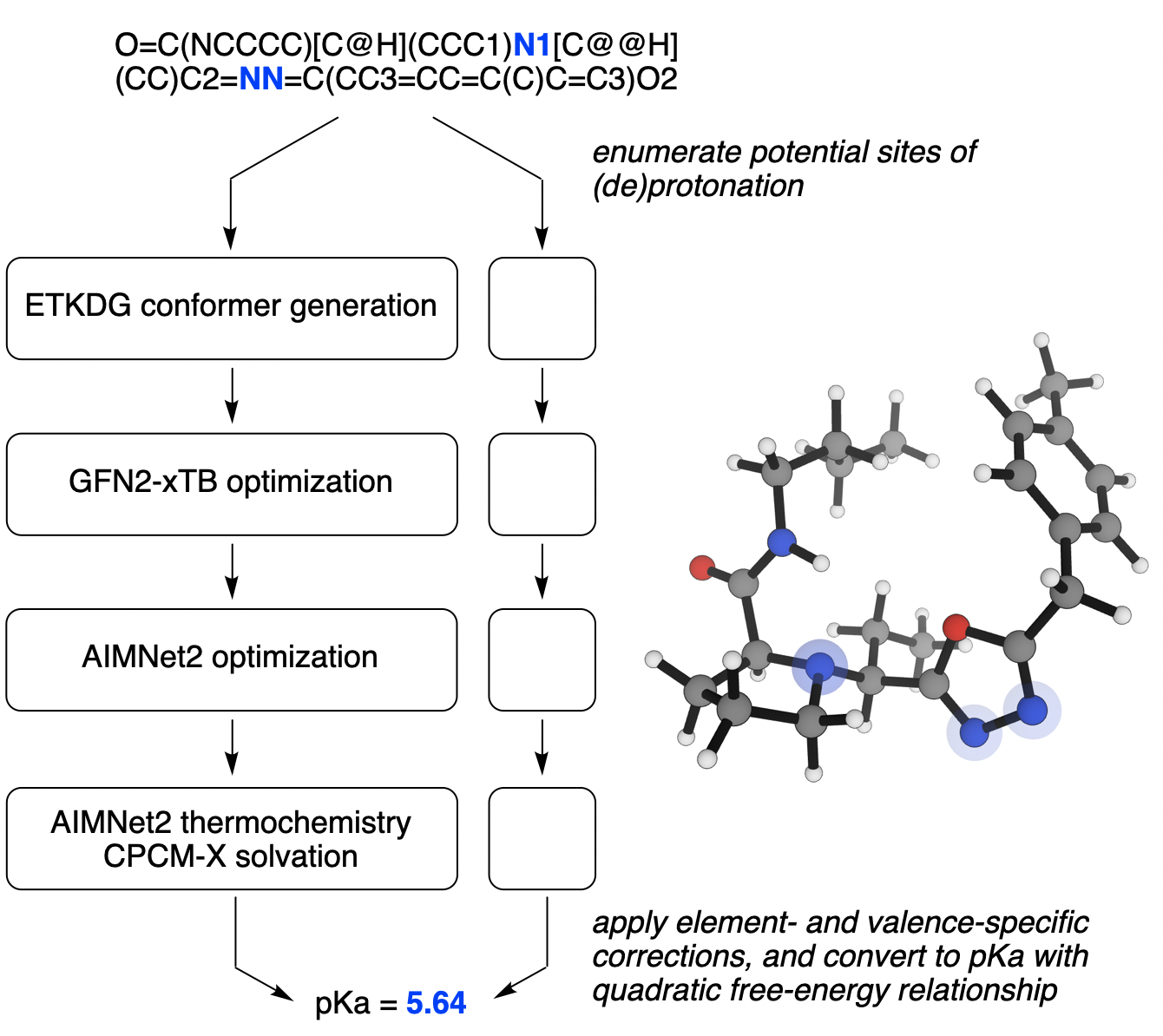
Figure 1. Visual overview of the Rowan pKa workflow.
Methodology
Our workflow begins by iteratively adding or removing hydrogens to the molecule of interest using RDKit and quickly estimating the proton affinity for the conjugate acid/conjugate base pair. If (de)protonation is predicted to yield a pKa value close to the desired range, conformers are generated using the ETKDG algorithm23,2423. Riniker, S.; Landrum, G. A. Better Informed Distance Geometry: Using What We Know To Improve Conformation Generation. J. Chem. Inf. Model.2015, 55, 2562–2574. 24. RDKit’s ETKDG algorithm consistently performs well in benchmarks, particularly when speed is at a premium: see (a) Friedrich, N.-O. et al. Benchmarking Commercial Conformer Ensemble Generators. J. Chem. Inf. Model.2017, 57, 2719–2728. (b) Ebejer, J.-P.; Morris, G. M.; Deane, C. M. Freely Available Conformer Generation Methods: How Good Are They? J. Chem. Inf. Model.2012, 52, 1146–1158. and optimized using the MMFF forcefield. After removing redundant geometries, low-energy conformers are then optimized using the GFN2-xTB semiempirical method,2525. Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB-An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput.2019, 15, 1652–1671. single-point energies are calculated using the AIMNet2 model trained on ωB97M-D3/def2-TZVPP (henceforth abbreviated as AN2-ωB97MD3),2222. Anstine, D. M.; Zubatyuk, R.; Isayev, O. AIMNet2: A Neural Network Potential to Meet your Neutral, Charged, Organic, and Elemental-Organic Needs. ChemRxiv, 2023, 10.26434/chemrxiv-2023-296ch. and the lowest N undergo full optimization and frequency calculations at the AN2-ωB97MD3 level of theory to generate a free energy for each conformer, including all standard entropy- and symmetry-based corrections.2626. Ochterski, J. W. Thermochemistry in Gaussian. Gaussian, 2022. https://gaussian.com/thermo/ (accessed Mar. 6, 2024). (N is set to 10 by default but can be varied by the end user; vide infra.) Since AIMNet2 does not take solvation into account, ∆Gsolv is computed from a single-point GFN2-xTB calculation with the CPCM-X implicit water model.2727. Stahn, M.; Ehlert, S.; Grimme, S. Extended Conductor-like Polarizable Continuum Solvation Model (CPCM-X) for Semiempirical Methods. J. Phys. Chem. A., 2023, 127, 7036–7043.
Following Pracht and Grimme, we combine energies from each conformer to generate a single hybrid ∆G for the conjugate acid/conjugate base pair and apply a quadratic free-energy relationship to convert this into pKa values.1818. Pracht, P.; Grimme, S. Efficient Quantum-Chemical Calculations of Acid Dissociation Constants from Free-Energy Relationships. J. Phys. Chem. A.2021, 125, 5681–5692. Initial application of this strategy provided disappointing results, with considerable systematic error observed between different classes of acids. Functional group-dependent systematic error is common in ab initio pKa prediction, and many different corrections have been employed: Schrödinger’s Jaguar pKa contains a different linear free-energy relationship for roughly a hundred different functional groups,2828. Bochevarov, A. D.; Watson, M. A.; Greenwood, J. R.; Philipp, D. M. Multiconformation, Density Functional Theory-Based pKa Prediction in Application to Large, Flexible Organic Molecules with Diverse Functional Groups. J. Chem. Theory Comput.2016, 12, 6001–6019. while Jensen and co-workers employed different reference molecules for each class of compound under study.1717. Kromann, J. C.; Larsen, F.; Moustafa, H.; Jensen, J. H. Prediction of pKa values using the PM6 semiempirical method. PeerJ, 2016, 4, e2335. To minimize the number of empirical parameters employed in our workflow, we instead add an element- and valence-specific constant to ∆G for each conjugate acid/conjugate base pair. (The correction for divalent oxygen, i.e. deprotonation of ROH, is defined as zero to eliminate extra degrees of freedom.) These constants are determined by Levenberg–Marquardt least-squares minimization, not by comparison to any individual reference compound, and can be thought of as a correction for systematic errors in both thermochemistry and solvation. Similar corrections are often used to account for dispersion in mean-field electronic structure theory methods without substantially compromising the generality of the method.2929. Grimme, S.; Hansen, A.; Brandenburg, J. G.; Bannwarth, C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev.2016, 116, 5105–5154.
Workflow development and parameter optimization was conducted on a slightly modified version of the TR224 dataset, also used by Pracht and Grimme.1818. Pracht, P.; Grimme, S. Efficient Quantum-Chemical Calculations of Acid Dissociation Constants from Free-Energy Relationships. J. Phys. Chem. A.2021, 125, 5681–5692. The final Rowan pKa workflow contains 7 tunable parameters, shown in Table 1.
Table 1. Tunable Parameters for Rowan pKa
Coefficients for Free-Energy Relationship
C0
-123.242550
C1
0.50677935 mol/kcal
C2
-1.7401e-04 mol2/kcal2
Element-/Valence-Specific Constants (kcal/mol)
N3
3.91398
N4
5.13054
C4
6.03872
O2
0.00 (defined)
S2
-5.14438
Results
Quantifying the accuracy of property prediction under realistic conditions is challenging: evaluation over a large dataset, while superficially appealing, often leads to exaggerated correlation coefficients that are not reproduced in smaller, less diverse populations.3030. Walters, P. We Need Better Benchmarks for Machine Learning in Drug Discovery. Practical Cheminformatics,2023. https://practicalcheminformatics.blogspot.com/2023/08/we-need-better-benchmarks-for-machine.html (accessed Mar. 6, 2024). Instead, we follow Rich and Birnbaum in using "assay-stratified metrics" to evaluate the predictive ability of our pKa workflow.3131. Rich, A.; Birnbaum, B. Get with the program. Inductive Bio,2023.https://www.inductive.bio/blog/building-better-benchmarks-for-adme-optimization (accessed Mar. 6, 2024). All datasets were scored using four metrics: mean absolute error (MAE), root mean square error (RMSE), the square of the Pearson correlation coefficient (r2), and Kendall’s tau (τ). Low values for MAE and RMSE reflect a high degree of absolute accuracy, while high values for r2 and τ demonstrate relative accuracy within a given dataset (arguably more predictive of performance under real-world usage).
SAMPL6 and SAMPL7 pKa Prediction Challenges
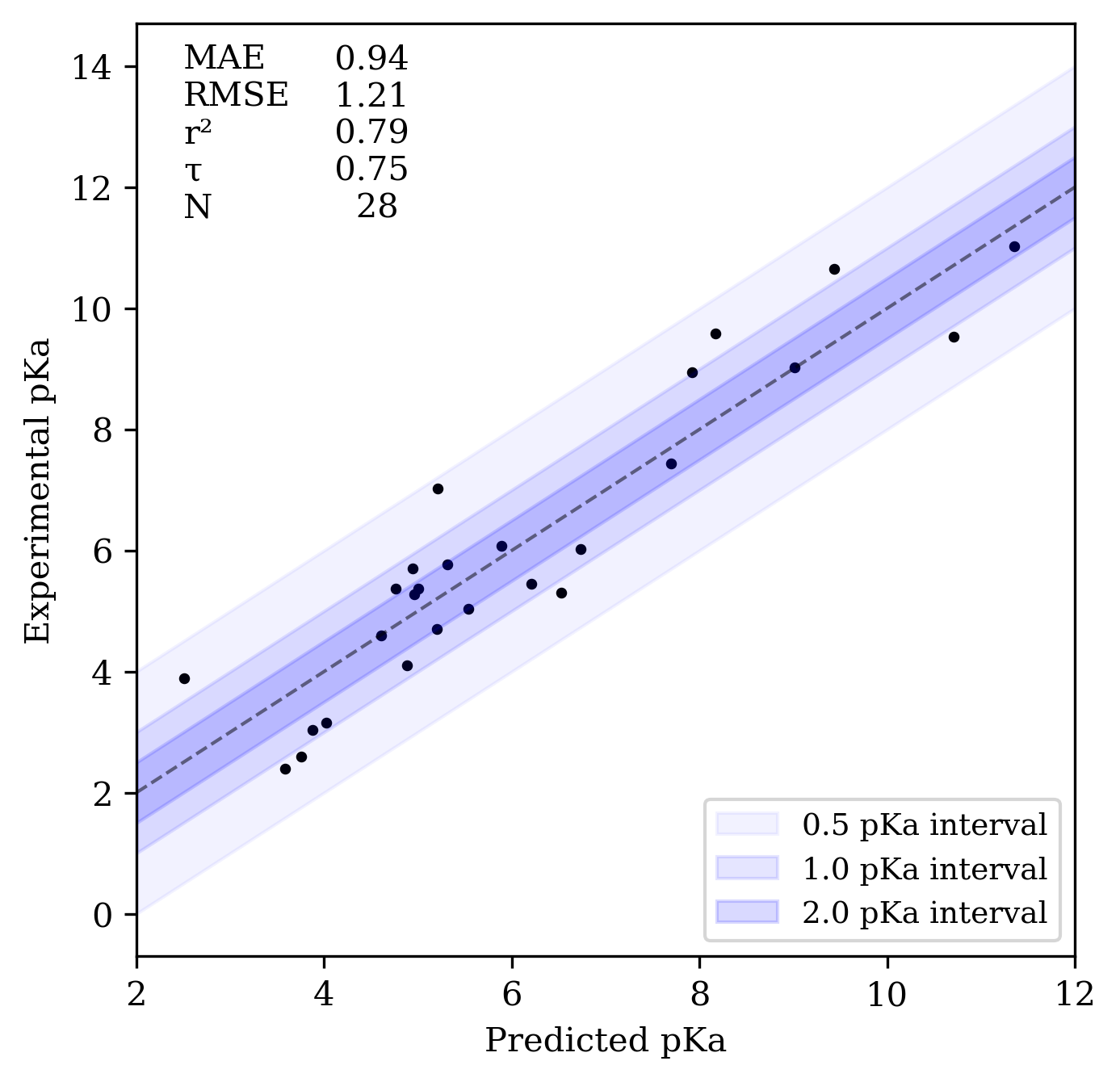
We began by evaluating Rowan pKa on datasets from the SAMPL6 and SAMPL7 blind pKa prediction challenges run several years ago. The SAMPL6 dataset consists of 24 molecules designed to resemble fragments of kinase inhibitors, many of which contain multiple potential sites of (de)protonation.1313. Işık, M. et al. Overview of the SAMPL6 pKa Challenge: Evaluating small molecule microscopic and macroscopic pKa predictions. J. Comput. Aided Mol. Des.2021, 35, 131–166. We computed microscopic pKavalues starting from each molecule’s neutral state and compared these values to the relevant macroscopic pKa values, excluding doubly ionized states. Rowan pKa performed well, with an MAE of 0.94 and an RMSE of 1.21, comparable to the performance of commercial pKaprediction tools like ChemAxon’s Chemicalize and Schrödinger’s Jaguar and Epik.3232. Direct comparison to SAMPL6 is complicated by the sophisticated statistical procedures conducted by the contest organizers (and by the fact that we exclude doubly ionized microstates from our analysis, making our SAMPL6 dataset subtly different from that studied by other methods), but at a high level the error metrics for Rowan pKa appear very comparable to those from the commercial programs mentioned in the text. The high values obtained for r2 and τ also illustrate the predictive utility of Rowan pKa, although the large range of pKa values in the SAMPL6 dataset makes obtaining a high value for r2 relatively easy.
Correlation plot for the SAMPL6 dataset.
Changes in Amine Basicity
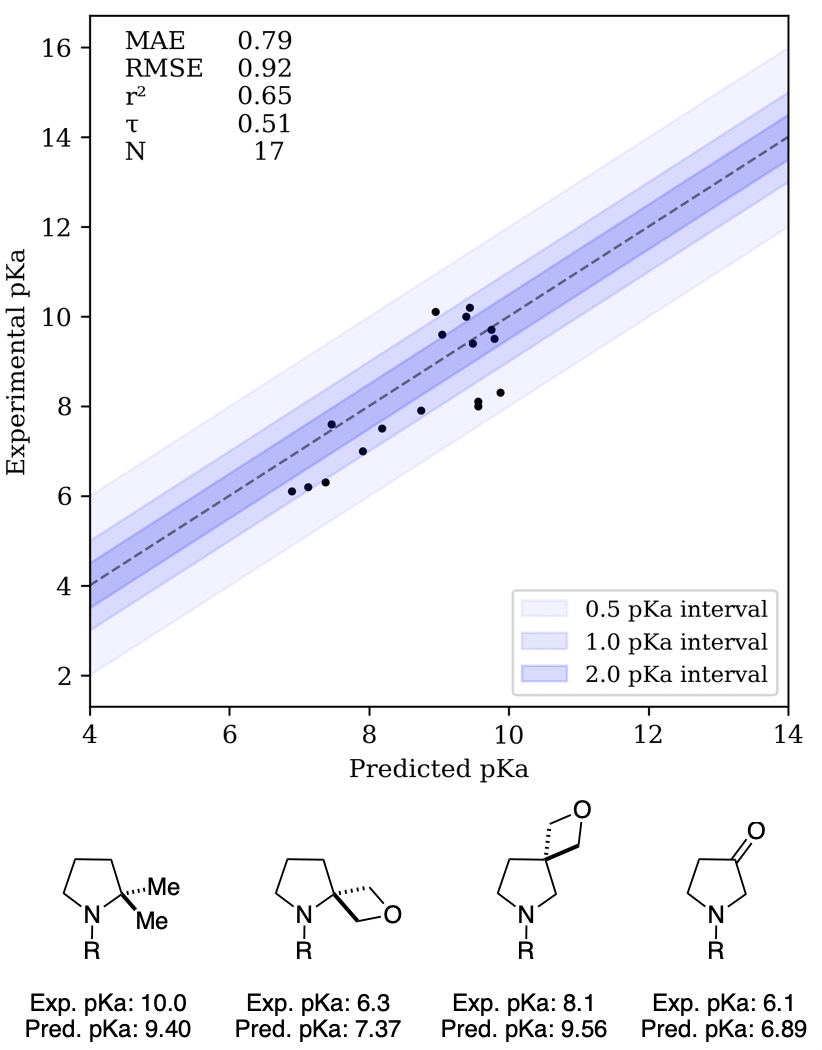
We next evaluated Rowan pKa on three datasets selected to showcase changes in amine basicity. In 2010, Müller and co-workers reported that introducing spiro-oxetane substituents in saturated N-heterocycles could attenuate the basicity of amines.3333. Wuitschik, G. et al. Oxetanes in Drug Discovery: Structural and Synthetic Insights. J. Med. Chem.2010,53, 3227–3246. We modeled their dataset using Rowan pKa, with the change that the bulky N-piperonyl group (added as a UV-absorbing handle for experimental convenience) was replaced with N-methyl in silico. Overall, good agreement between experimental and predicted pKa was observed, although the effect of β-oxetane substituents was systematically underestimated.
Correlation plot for the Müller spiro-oxetane dataset and illustration of Rowan’s failure to model β-oxetane substituents (R = piperonyl for experiments, methyl for computations).
pKa Values from Medicinal Chemistry Literature
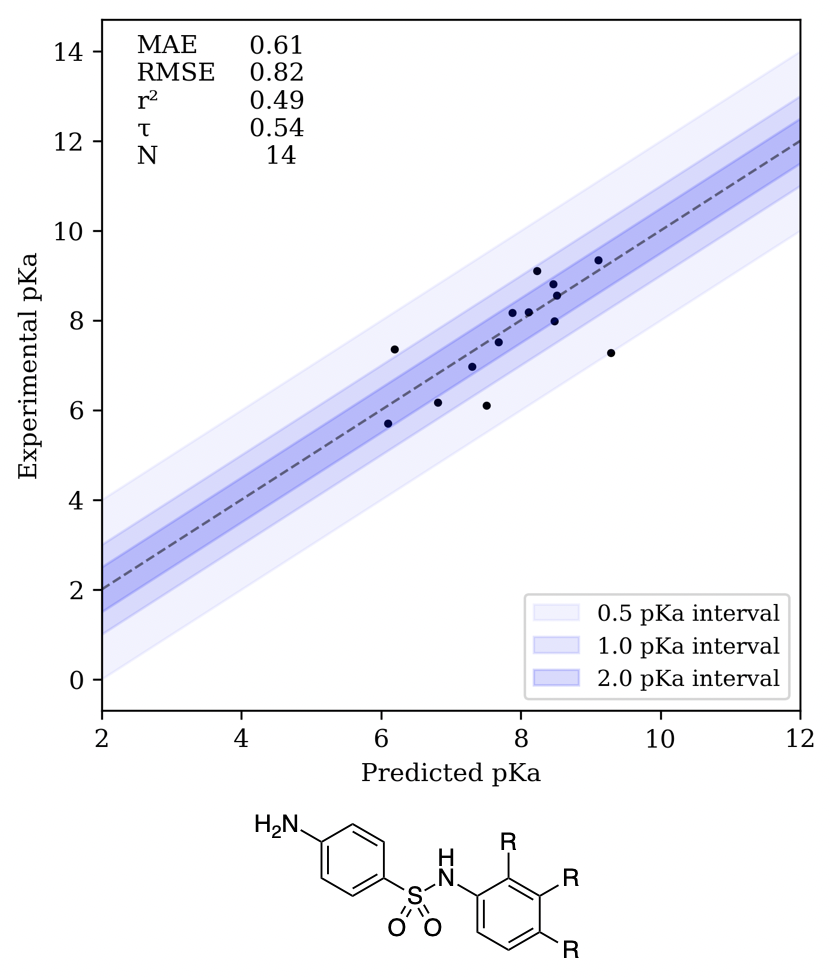
We next examined the ability of Rowan to recapitulate pKa values from the medicinal chemistry literature. In 1972, Miller, Doukas, and Seydel reported that N-aryl sulfonamides served as potent inhibitors of folate synthesis and found that the inhibitory activity could be correlated with the pKa of the sulfonamide N–H.3636. Miller, G. H.; Doukas, P. H.; Seydel, J. K. Sulfonamide structure-activity relationship in a cell-free system. Correlation of inhibition of folate synthesis with antibacterial activity and physicochemical parameters. J. Med. Chem.1972,15, 700–706. The Rowan pKa workflow recapitulates the experimental trend in pKa values and thus also the observed trend in potency, illustrating the utility of pKa calculations in this system.
Correlation plot for the folate dataset and illustration of the sulfonamides investigated by Miller, Doukas, and Seydel.
Macrocycles
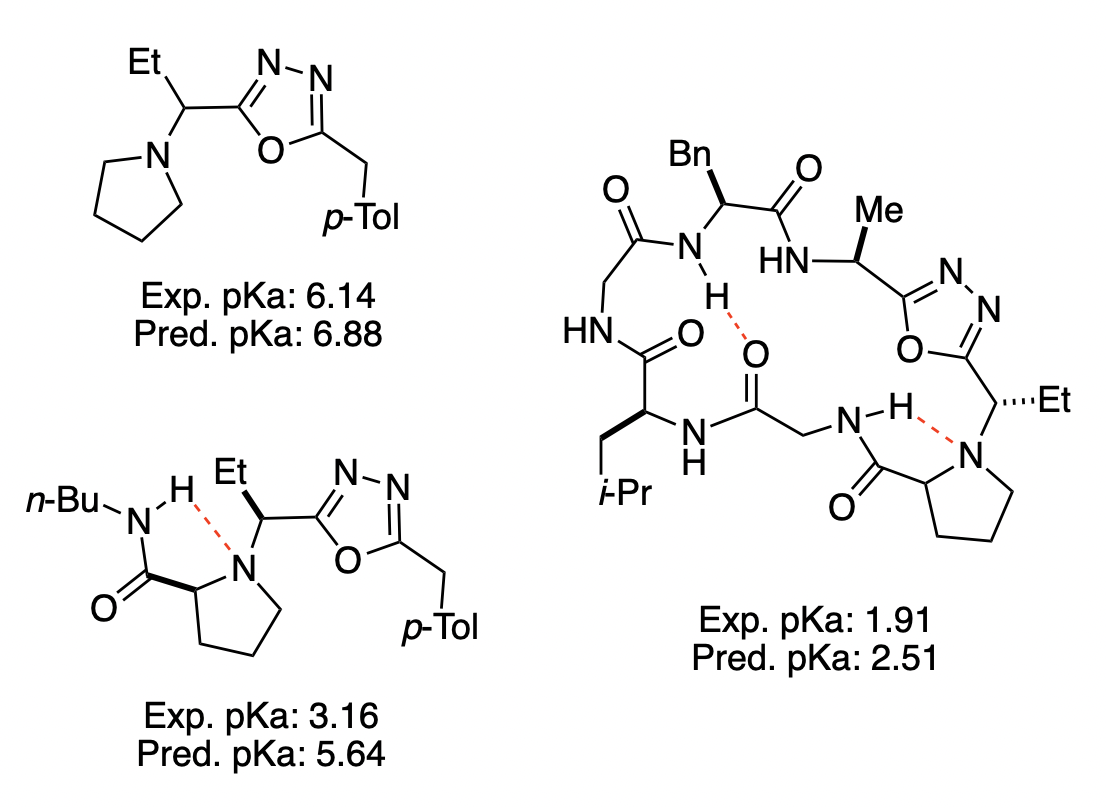
We next examined the ability of Rowan pKa to predict the pKa of macrocyclic peptides. Macrocyclic peptides inhabit different conformations from their linear congeners and frequently possess markedly different pKa values, with significant consequences for solubility and bioavailability. In 2018, Yudin and co-workers reported that the pKa of reduced amide bonds adjacent to oxadiazole rings was lowered in macrocycles, which they attributed to intramolecular hydrogen bonding that stabilizes the neutral amine.4040. Appavoo, S. D. et al. Development of Endocyclic Control Elements for Peptide Macrocycles. J. Am. Chem. Soc.2018,140, 8763–8770. Rowan pKa was able to recapitulate the observed trend in pKa values (Figure 10), which would be considerably more challenging for QSPR-type methods that do not explicitly consider conformational effects.
Figure 10. Comparison of predicted and experimentally measured pKa values for various oxadiazole/pyrrolidine-containing structures, with the intramolecular hydrogen bonds proposed by Yudin and co-workers illustrated in red.
High–Throughput pKa Prediction
Finally, we explored the suitability of Rowan pKa as a high-throughput method for pKaprediction. We selected the first 100 rings from Peter Ertl’s recently published library of medicinal chemistry-relevant ring systems as a test set (15.5 atoms/molecule on average) and investigated whether the accuracy of the conformation search/refinement process could be further reduced to increase throughput without significantly hurting the workflow’s accuracy.4141. Ertl, P. Database of 4 Million Medicinal Chemistry-Relevant Ring Systems. J. Chem. Inf. Model.2024,64, 1245–1250. Two modes with reduced conformer searching were eventually developed: the key changes made for each new mode are summarized in Table 2. Modest gains in efficiency are possible, but at the cost of significantly increased error: if multiple low-energy conformers exist, these data suggest that they should be included.4242. These results contrast with Bochevarov and co-workers’ finding (ref. 28) that decent results can be obtained from only a single conformer—the rapid workflows here may prune conformers too aggressively and often fail to find the global minimum. While electronic structure theory-based approaches can easily spend several minutes refining conformers without substantially impacting the overall runtime, the efficiency of AIMNet2 makes fast conformational searching crucial for speeding pKa prediction.
Table 2. Different Modes for Conformer Searching
Parameter
Careful
Rapid
Reckless
# initial confs
250
100
50
initial energy cutoff (kcal/mol)
15.0
10.0
5.0
RMSD threshold (Å)
0.10
0.25
0.50
# confs. (xTB)
20
10
3
final energy cutoff (kcal/mol)
5.0
5.0
3.0
# confs. (AIMNet2)
10
3
1
final energy cutoff (kcal/mol)
15.0
10.0
5.0
MAE (relative to "Careful")
0.00
0.38
0.69
RMSE (relative to "Careful")
0.00
1.17
1.66
Average Time (s)a
23.05
16.01
14.13
aTimings reported for an EC2 c5.2xlarge instance.
Discussion
In this work, we show that relatively aggressive approximations can be applied to "gold standard" approaches like Grimme’s xvxzd workflow while still maintaining useful accuracy. The xvxzd workflow consists of extensive metadynamics-based conformation searching in crest, geometry optimization with PBEh-3c in implicit solvent, single-point energy calculations using the double-hybrid functional DSD-BLYP-D3(BJ)/def2-TZVPD, and subsequent implicit solvent calculations using COSMO-RS; here, we replace crestwith RDKIT’s ETKDG conformer generation algorithm, DSD-BLYP-D3(BJ) with AIMNet2, and COSMO-RS with CPCM-X.1515. Pracht, P. et al. High accuracy quantum-chemistry-based calculation and blind prediction of macroscopic pKa values in the context of the SAMPL6 challenge. J. Comput. Aided Mol. Des.2018, 32, 1139–1149. While Rowan pKa is not as fast as QSPR-based approaches or as accurate as high-level electronic structure theory-based approaches like xvxzd, the combination of accuracy, speed, and minimal empiricism achieved by our workflow is nevertheless appealing. We anticipate that the ability to obtain fast and reasonable pKa values for unseen regions of chemical space will be particularly useful in drug discovery. Owing to the lack of functional group-specific corrections, the pKa values computed by Rowan are ideally suited to downstream manipulation or scaling, which can further reduce systematic error where desired.
What limits the accuracy of the Rowan pKa workflow, and what can be done to improve it? At a high level, errors in ab initio microscopic pKa prediction can come from several different sources: (1) insufficient consideration of conformational effects or analysis of the wrong conformations, (2) poor description of the underlying gas-phase thermochemistry, or (3) inaccurate description of solvation. To achieve maximum efficiency, a Pareto-optimal pKaprediction workflow ought to balance errors from all three sources without "overspending" for a high-accuracy answer in only one area. In specific cases, we have been able to track down anomalous predictions made by Rowan pKato all three underlying potential sources of error—conformational searching, gas phase thermochemistry, and implicit solvent model—indicating that no factor is solely responsible for the observed error. While improvements to conformational generation4444. Jing, B. et al. Torsional Diffusion for Molecular Conformer Generation. arXiv,2023, 10.48550/arXiv:2206.01729. and machine-learned interatomic potentials4545. Liao, Y.-L.; Wood, B.; Das, A.; Smidt, T. EquiformerV2: Improved Equivariant Transformer for Scaling to Higher-Degree Representations. arXiv,2023, 10.48550/arXiv.2306.12059. are easy to envision based on current research, improvements to implicit solvation may prove to be more elusive: despite repeated calls for alternatives to "scandalous" implicit solvation,4646. Grimme, S.; Schreiner, P. R. Computational Chemistry: The Fate of Current Methods and Future Challenges. Angew. Chem. Int. Ed.2017,57, 4170–4176. no practical and general alternatives have yet been developed. Recent work in coarse-grained machine-learned potentials may point towards a new paradigm for handling solvent effects, but more work is needed to investigate the strengths and limitations of such approaches.48,4948. Arts, M. et al. Two for One: Diffusion Models and Force Fields for Coarse-Grained Molecular Dynamics. J. Chem. Theory Comput.2023,19, 6151–6159. 49. Zhang, J.; Pagotto, J.; Gould, T.; Duignan, T. T. Accurate, fast and generalizable first principles simulation of aqueous lithium chloride. arXiv,2023, 10.48550/arXiv.2310.12535.
More generally, the approach described in this paper divides the task of predicting pKainto two separate components: (1) a general pre-trained model which maps molecular structures to energies, thus "learning chemistry" at a high level, and (2) an application-specific workflow which translates geometries and energies generated by the pre-trained model into predictions relevant for the task at hand. This partitioning has certain advantages: the general pre-trained model (here AIMNet2) is systematically improvable and can be trained using purely in silicodata, in principle allowing arbitrarily high levels of accuracy to be attained, while the application-specific workflow relies largely on conventional computational chemistry techniques and does not require extensive experimental training data like ML-based QSPR methods. As the accuracy of machine-learned interatomic potentials improves, we anticipate that the strategy demonstrated here—using machine-learned interatomic potentials as drop-in replacements for quantum chemistry—will become a general strategy for accelerating computational chemistry.
To run this pKa prediction workflow on your own datasets and structures, create an account on Rowan today. All new users get 500 free credits, enough to run hundreds of pKa predictions.
Acknowledgment
The authors acknowledge Abba Leffler, Diego Diaz, Eugene Kwan, Joe Gair, Pat Walters, and Philipp Pracht for helpful discussions.
References
Di, L.; Kerns, E. H. Drug-Like Properties: Concepts, Structure Design, and Methods, 2nd ed.; Academic Press, 2016.
Charifson, P. S.; Walters, W. P. Acidic and Basic Drugs in Medicinal Chemistry: A Perspective. J. Med. Chem.2014, 57, 9701–9717.
Garrido, A. et al. hERG toxicity assessment: Useful guidelines for drug design. Eur. J. Med. Chem.2020, 195, 112290.
Ploeman, J.-P. H.T.M. et al. Use of physicochemical calculation of pKa and CLogP to predict phospholipidosis-inducing potential: A case study with structurally related piperazines. Exp. Toxicol. Pathol.2004, 55, 347–355.
Pelletier, D. J. et al. Evaluation of a Published in Silico Model and Construction of a Novel Bayesian Model for Predicting Phospholipidosis Inducing Potential. J. Chem. Inf. Model.2007, 47, 1196–1205.
Manallack, D. T. The pKa Distribution of Drugs: Application to Drug Discovery. Perspect. Medicin. Chem.2007, 1, 25–38.
Meloun, M.; Bordovská, S. Benchmarking and validating algorithms that estimate pKa values of drugs based on their molecular structures. Anal. Bioanal. Chem.2007, 389, 1267–1281.
Liao, C.; Nicklaus, M. C. Comparison of Nine Programs Predicting pKa Values of Pharmaceutical Substances. J. Chem. Inf. Model.2009, 49, 2801–2812.
Settimo, L.; Bellman, K.; Knegtel, R. M. A. Comparison of the Accuracy of Experimental and Predicted pKa Values of Basic and Acidic Compounds. Pharm. Res.2013, 31, 1082–1095.
For state-of-the-art QSPR methods, see: (a) Johnston, R. C. et al. Epik: pKa and Protonation State Prediction through Machine Learning. ChemRxiv, 2023, 10.26434/chemrxiv-2023-c6z8t-v3. (b) Mayr, F.; Wieder, M.; Wieder, O.; Langer, T. Improving Small Molecule pKa Prediction Using Transfer Learning With Graph Neural Networks. Front. Chem.2022, 10, 866585. (c) Luo, W. et al. Uni-pKa: An Accurate and Physically Consistent pKa Prediction through Protonation Ensemble Modeling. ChemRxiv, 2023, 10.26434/chemrxiv-2023-lw5k0.
Rupp, M.; Körner, R.; Tetko, I. V. Predicting the pKa of Small Molecules. Comb. Chem. High Throughput Screen.2011, 14, 307–327.
Navo, C. D.; Jiménez-Osés, G. Computer Prediction of pKa Values in Small Molecules and Proteins. ACS Med. Chem. Lett.2021, 12, 1624–1628.
Işık, M. et al. Overview of the SAMPL6 pKa Challenge: Evaluating small molecule microscopic and macroscopic pKa predictions. J. Comput. Aided Mol. Des.2021, 35, 131–166.
Bergazin, T. D. et al. Evaluation of log P, pKa, and log D predictions from the SAMPL7 blind challenge. J. Comput. Aided Mol. Des.2021, 35, 771–802.
Pracht, P. et al. High accuracy quantum-chemistry-based calculation and blind prediction of macroscopic pKa values in the context of the SAMPL6 challenge. J. Comput. Aided Mol. Des.2018, 32, 1139–1149.
Based on current AWS EC2 on-demand pricing (Feb. 26, 2024) a c5.4xlarge instance with 16 vCPUs costs $0.68/hour: $0.68/hour * 28/16 * 24 hours = $28.56.
Kromann, J. C.; Larsen, F.; Moustafa, H.; Jensen, J. H. Prediction of pKa values using the PM6 semiempirical method. PeerJ, 2016, 4, e2335.
Pracht, P.; Grimme, S. Efficient Quantum-Chemical Calculations of Acid Dissociation Constants from Free-Energy Relationships. J. Phys. Chem. A.2021, 125, 5681–5692.
Alongi, K. S.; Shields, G. C. Theoretical Calculations of Acid Dissociation Constants: A Review Article. In Annual Reports in Computational Chemistry,; Wheeler, R. A., Ed.; Elsevier, 2010; Vol. 6, 113–138.
Dékány, A. A.; Czakó, G. Benchmark ab initio proton affinity and gas-phase basicity of α-alanine based on coupled-cluster theory and statistical mechanics. J. Comput. Chem.2021, 43, 19–28.
See, inter alia, (a) Behler, J. Four Generations of High-Dimensional Neural Network Potentials. Chem. Rev.2021, 121, 10037–10072. (b) Friederich, P.; Häse, F.; Proppe, J.; Aspuru-Guzik, A. Machine-learned potentials for next-generation matter simulations. Nat. Mater.2021, 20, 750–761. (c) Anstine, D. M.; Isayev, O. Machine Learning Interatomic Potentials and Long-Range Physics. J. Phys. Chem. A.2023, 127, 2417–2431.
Anstine, D. M.; Zubatyuk, R.; Isayev, O. AIMNet2: A Neural Network Potential to Meet your Neutral, Charged, Organic, and Elemental-Organic Needs. ChemRxiv, 2023, 10.26434/chemrxiv-2023-296ch.
Riniker, S.; Landrum, G. A. Better Informed Distance Geometry: Using What We Know To Improve Conformation Generation. J. Chem. Inf. Model.2015, 55, 2562–2574.
RDKit’s ETKDG algorithm consistently performs well in benchmarks, particularly when speed is at a premium: see (a) Friedrich, N.-O. et al. Benchmarking Commercial Conformer Ensemble Generators. J. Chem. Inf. Model.2017, 57, 2719–2728. (b) Ebejer, J.-P.; Morris, G. M.; Deane, C. M. Freely Available Conformer Generation Methods: How Good Are They? J. Chem. Inf. Model.2012, 52, 1146–1158.
Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB-An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput.2019, 15, 1652–1671.
Ochterski, J. W. Thermochemistry in Gaussian. Gaussian, 2022. https://gaussian.com/thermo/ (accessed Mar. 6, 2024).
Stahn, M.; Ehlert, S.; Grimme, S. Extended Conductor-like Polarizable Continuum Solvation Model (CPCM-X) for Semiempirical Methods. J. Phys. Chem. A., 2023, 127, 7036–7043.
Bochevarov, A. D.; Watson, M. A.; Greenwood, J. R.; Philipp, D. M. Multiconformation, Density Functional Theory-Based pKa Prediction in Application to Large, Flexible Organic Molecules with Diverse Functional Groups. J. Chem. Theory Comput.2016, 12, 6001–6019.
Grimme, S.; Hansen, A.; Brandenburg, J. G.; Bannwarth, C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev.2016, 116, 5105–5154.
Direct comparison to SAMPL6 is complicated by the sophisticated statistical procedures conducted by the contest organizers (and by the fact that we exclude doubly ionized microstates from our analysis, making our SAMPL6 dataset subtly different from that studied by other methods), but at a high level the error metrics for Rowan pKa appear very comparable to those from the commercial programs mentioned in the text.
Wuitschik, G. et al. Oxetanes in Drug Discovery: Structural and Synthetic Insights. J. Med. Chem.2010,53, 3227–3246.
Smyrnov, O. et al. α-CF3-Substituted Saturated Bicyclic Amines: Advanced Building Blocks for Medicinal Chemistry. Eur. J. Org. Chem.2024,27, e202300935.
Lõkov, M. et al. On the Basicity of Conjugated Nitrogen Heterocycles in Different Media. Eur. J. Org. Chem.2017,30, 4475–4489.
Miller, G. H.; Doukas, P. H.; Seydel, J. K. Sulfonamide structure-activity relationship in a cell-free system. Correlation of inhibition of folate synthesis with antibacterial activity and physicochemical parameters. J. Med. Chem.1972,15, 700–706.
Oehlrich, D.; Prokopcova, H.; Gijsen, H. J. M. The evolution of amidine-based brain penetrant BACE1 inhibitors. Bioorg. Med. Chem. Lett.2014,24, 2033–2045.
Rombouts, F. J. R. et al. Modulating physicochemical properties of tetrahydropyridine-2-amine BACE1 inhibitors with electron-withdrawing groups: A systematic study. Eur. J. Med. Chem.2022,228, 114028.
Morgenthaler, M. et al. Predicting and Tuning Physicochemical Properties in Lead Optimization: Amine Basicities. ChemMedChem,2007,2, 1100–1115.
Appavoo, S. D. et al. Development of Endocyclic Control Elements for Peptide Macrocycles. J. Am. Chem. Soc.2018,140, 8763–8770.
Ertl, P. Database of 4 Million Medicinal Chemistry-Relevant Ring Systems. J. Chem. Inf. Model.2024,64, 1245–1250.
These results contrast with Bochevarov and co-workers’ finding (ref. 28) that decent results can be obtained from only a single conformer—the rapid workflows here may prune conformers too aggressively and often fail to find the global minimum. While electronic structure theory-based approaches can easily spend several minutes refining conformers without substantially impacting the overall runtime, the efficiency of AIMNet2 makes fast conformational searching crucial for speeding pKa prediction.
Implicit solvation has been demonstrated to be a leading source of error for conventional electronic structure theory-based pKa prediction. See, inter alia, (a) Thapa, B.; Schlegel, H. B. Density Functional Theory Calculation of pKa’s of Thiols in Aqueous Solution Using Explicit Water Molecules and the Polarizable Continuum Model. J. Phys. Chem. A2016,120, 5726–5735. (b) Thapa, B.; Raghavachari, K. Accurate pKa Evaluations for Complex Bio-Organic Molecules in Aqueous Media. J. Chem. Theory Comput.2019,15, 6025–6035.
Jing, B. et al. Torsional Diffusion for Molecular Conformer Generation. arXiv,2023, 10.48550/arXiv:2206.01729.
Liao, Y.-L.; Wood, B.; Das, A.; Smidt, T. EquiformerV2: Improved Equivariant Transformer for Scaling to Higher-Degree Representations. arXiv,2023, 10.48550/arXiv.2306.12059.
Grimme, S.; Schreiner, P. R. Computational Chemistry: The Fate of Current Methods and Future Challenges. Angew. Chem. Int. Ed.2017,57, 4170–4176.
Cluster–continuum solvation shows promise but is difficult to run in a black-box manner and introduces considerable additional complexity. See (a) Pliego, J. R.; Riveros, J. M. The Cluster−Continuum Model for the Calculation of the Solvation Free Energy of Ionic Species. J. Phys. Chem. A2001,105, 7241–7247. (b) Norjmaa, G.; Ujaque, G.; Lledós, A. Beyond Continuum Solvent Models in Computational Homogeneous Catalysis. Top. Catal.2022,65, 118–140. (c) Spicher, S. et al. Automated Molecular Cluster Growing for Explicit Solvation by Efficient Force Field and Tight Binding Methods. J. Chem. Theory Comput.2022,18, 3174–3189.
Arts, M. et al. Two for One: Diffusion Models and Force Fields for Coarse-Grained Molecular Dynamics. J. Chem. Theory Comput.2023,19, 6151–6159.
Zhang, J.; Pagotto, J.; Gould, T.; Duignan, T. T. Accurate, fast and generalizable first principles simulation of aqueous lithium chloride. arXiv,2023, 10.48550/arXiv.2310.12535.