End-to-End Protein–Ligand Cofolding with Boltz-1
For a comparison against Rowan's other co-folding models, see our co-folding overview page.
Most computational docking workflows assume a fixed protein conformation, placing ligands into a rigid binding site. This approach is efficient but can fail when binding depends on structural rearrangements in the protein. Side-chain rotations, loop movements, and backbone shifts are common in experimentally determined complexes, yet many in silico pipelines either ignore these effects or treat them only partially.
Here's an image of what this ligand-induced reorganization can look like: the apo protein structure is shown in magenta, while the predicted holo structure is shown in cyan.
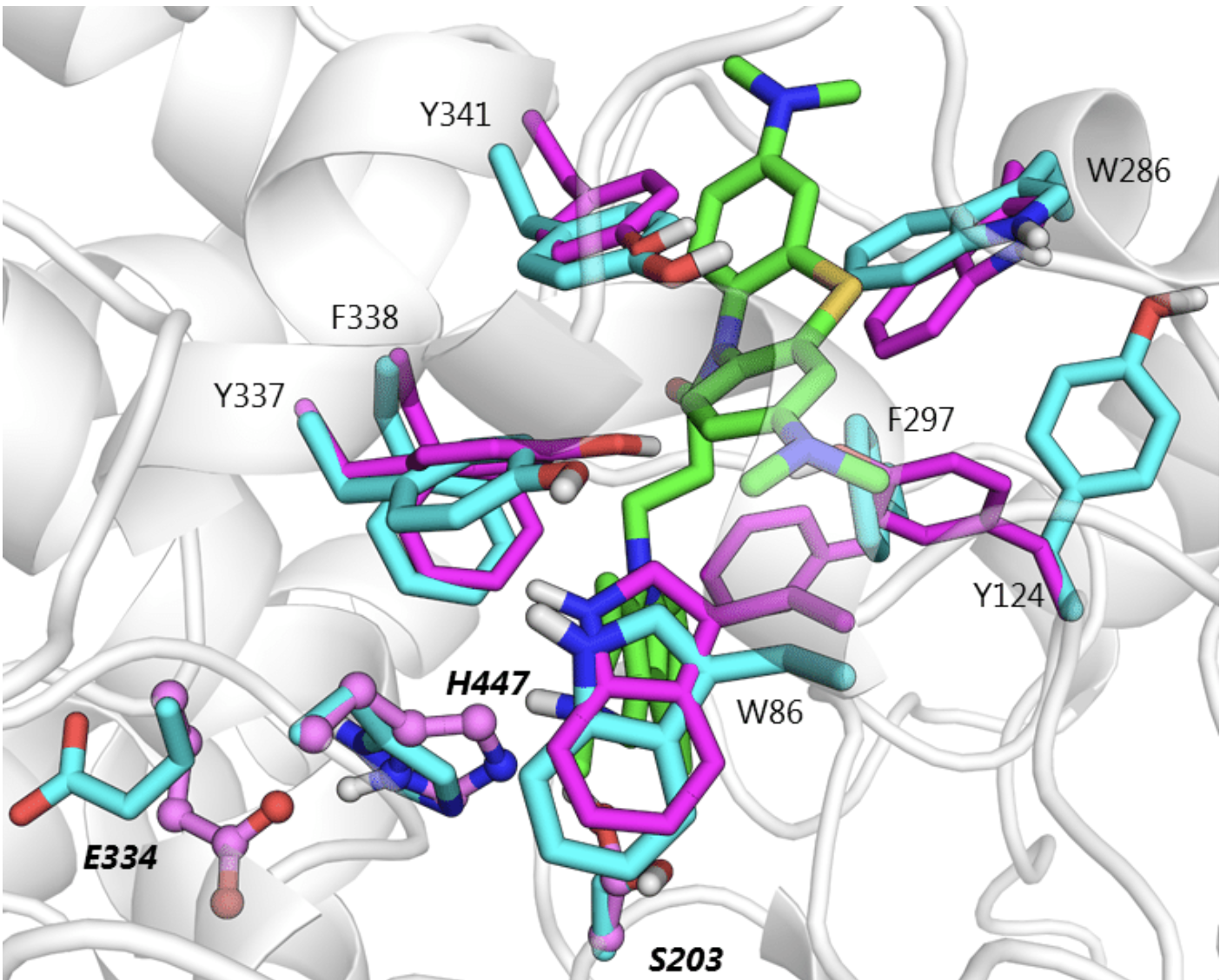
Comparison of apo (magenta) and predicted holo (cyan) protein structure
Boltz-1 uses a diffusion-based generative model that predicts protein and ligand structures together in their bound state. Rather than inserting a ligand into a static receptor, the model learns a joint distribution over both molecules' conformations. This makes it possible to capture realistic induced-fit geometry and to propose binding poses that incorporate flexibility in both partners.
This page does not cover the newer Boltz-2 or Boltz-2.1 models—for these methods, see our Boltz-2 page.
Method Overview
Boltz-1 is built on an SE(3)-equivariant diffusion architecture trained on experimentally resolved protein–ligand complexes from the Protein Data Bank. The generation process allows both side-chain and backbone atoms to move during prediction, so binding-site rearrangements can be captured without additional refinement steps. For each prediction, Boltz-1 reports two confidence metrics: the interface TM-score (iTM), which quantifies the structural agreement at the binding interface, and the predicted local distance difference test (pLDDT), which estimates per-residue accuracy.
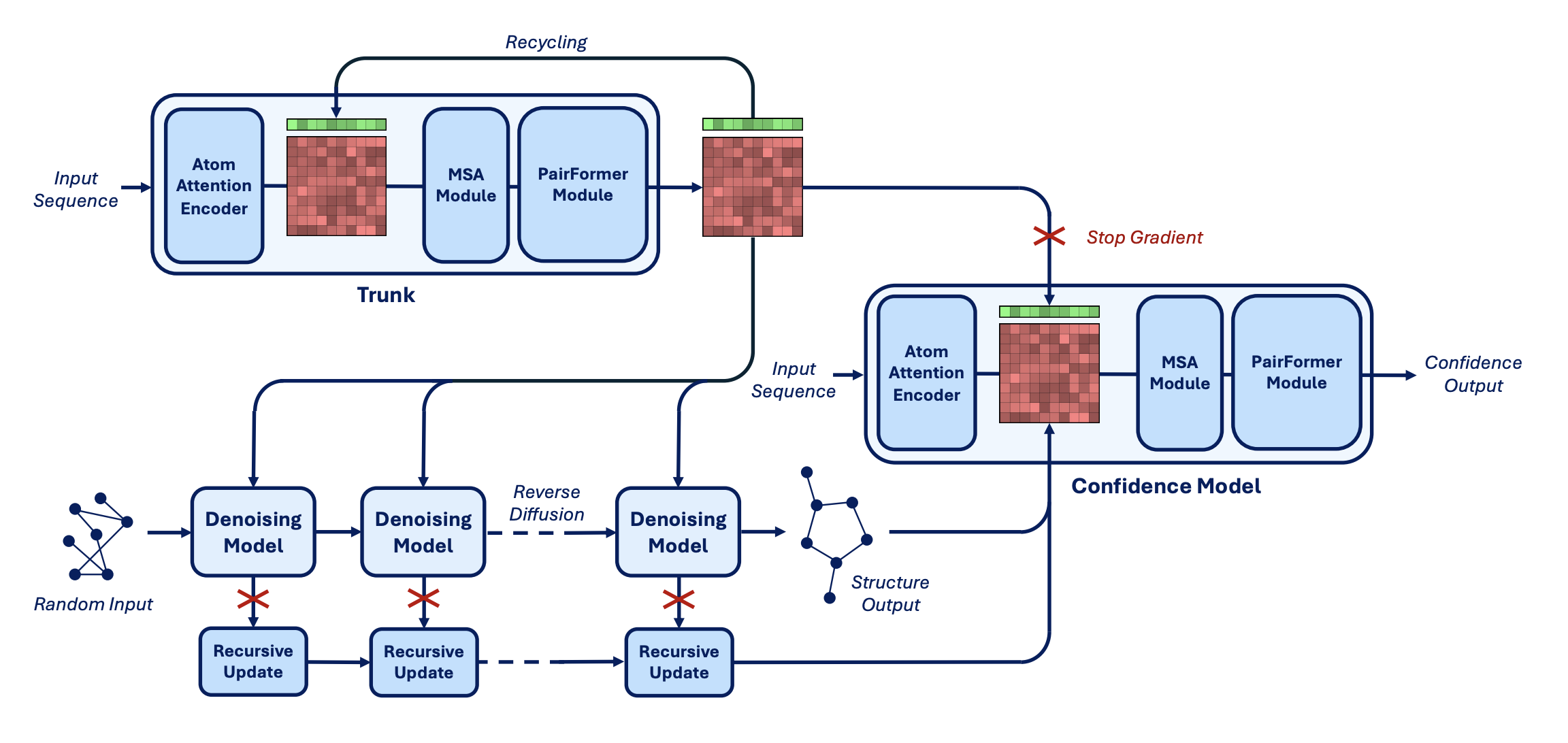
The architecture of Boltz-1 (Figure 3 from the preprint).
For full methodological details, please refer to the original publication by Jeremy Wohlwend and co-workers.
Performance
There are a growing number of co-folding methods—AlphaFold 3, Boltz-1, Chai-1, and others—which naturally raises the question of how these models compare. Many benchmarks have been run, most of which find that performance is very comparable between the three models. Here's Brian Naughton's benchmark conclusions:
On my antibody benchmark, Boltz and Chai-1 perform eerily similar, with a couple of cases where Boltz wins out. That, combined with all the data from the literature, makes the conclusion straightforward, at least for me. Boltz performs as well as or better than any of the models, has a clean, complete codebase with relatively little code, is hackable, and is by far the most open model. I am excited to see how Boltz progresses in 2025!
Still, a word of caution is warranted. Results from Peter Škrinjar and co-workers show that the performance of protein–ligand co-folding methods can be predicted almost perfectly by similarity to the training set, which suggests that these models are not in fact capable of extrapolation to unusual unseen systems.
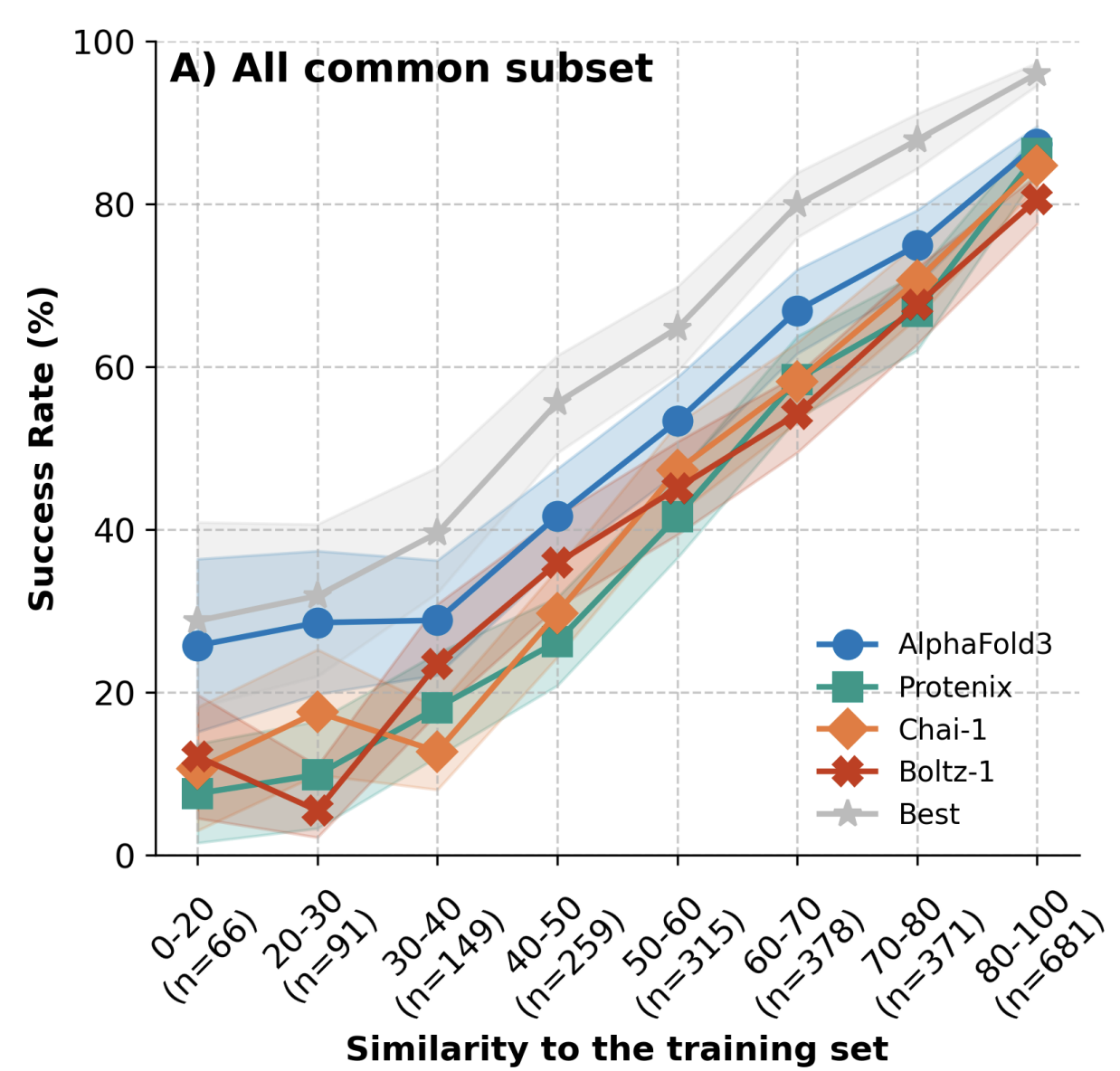
Similarity to the training set predicts cofolding performance.
(On Practical Cheminformatics, Pat Walters discusses this paper and a variety of other studies demonstrating that Boltz-1 and related methods don't yet understand binding physics correctly.)
Applications
Boltz-1 can be used to generate hypotheses about induced-fit binding mechanisms before undertaking more computationally intensive molecular dynamics simulations. It is well suited to exploring ligand binding modes in targets with cryptic or flexible sites, and it can support early-stage discovery when no holo crystal structure is available. In each of these cases, the method provides both a predicted structure and quantitative measures of its reliability.
Here's what Pat Walters has to say about the current use of co-folding methods in drug discovery:
I have been using co-folding methods in a similar way to how I use molecule generation techniques— as tools to develop hypotheses that can be tested experimentally. We have many methods that tell us if a molecule binds, such as biochemical assays, size exclusion chromatography-MS, examining binding in a DEL screen, or performing various other experiments. However, apart from X-ray crystallography or cryoEM, we have very few experiments that can definitively identify where a compound binds. In most cases, we need a hypothesis to guide experiments, such as mutagenesis or installing a photoaffinity label. Even more definitive experiments, like NMR or HDX-MS, are much more useful when a binding hypothesis is available. Co-folding provides an easy way to generate these hypotheses; then, it's up to our scientific knowledge, intuition, and creativity to decide how to proceed.
This conclusion—that co-folding methods are promising hypothesis generators but no substitute for experimental data—seems to match what we're seeing from lots of other drug-discovery teams.
Accessing Boltz-1 in Rowan
GUI-Based Submission
The Rowan platform includes an implementation of Boltz-1 that can be run directly in a web browser. Users upload their protein and ligand structures, initiate the cofolding procedure, and receive bound-state predictions complete with confidence metrics.
An interactive 3D viewer is provided for inspection of binding interactions, hydrogen bonding, and other contact features. This removes the need for local installation or specialized hardware, allowing researchers to focus on interpreting results rather than managing compute resources.
Here's what the result of a Boltz-1 calculation looks like on Rowan:
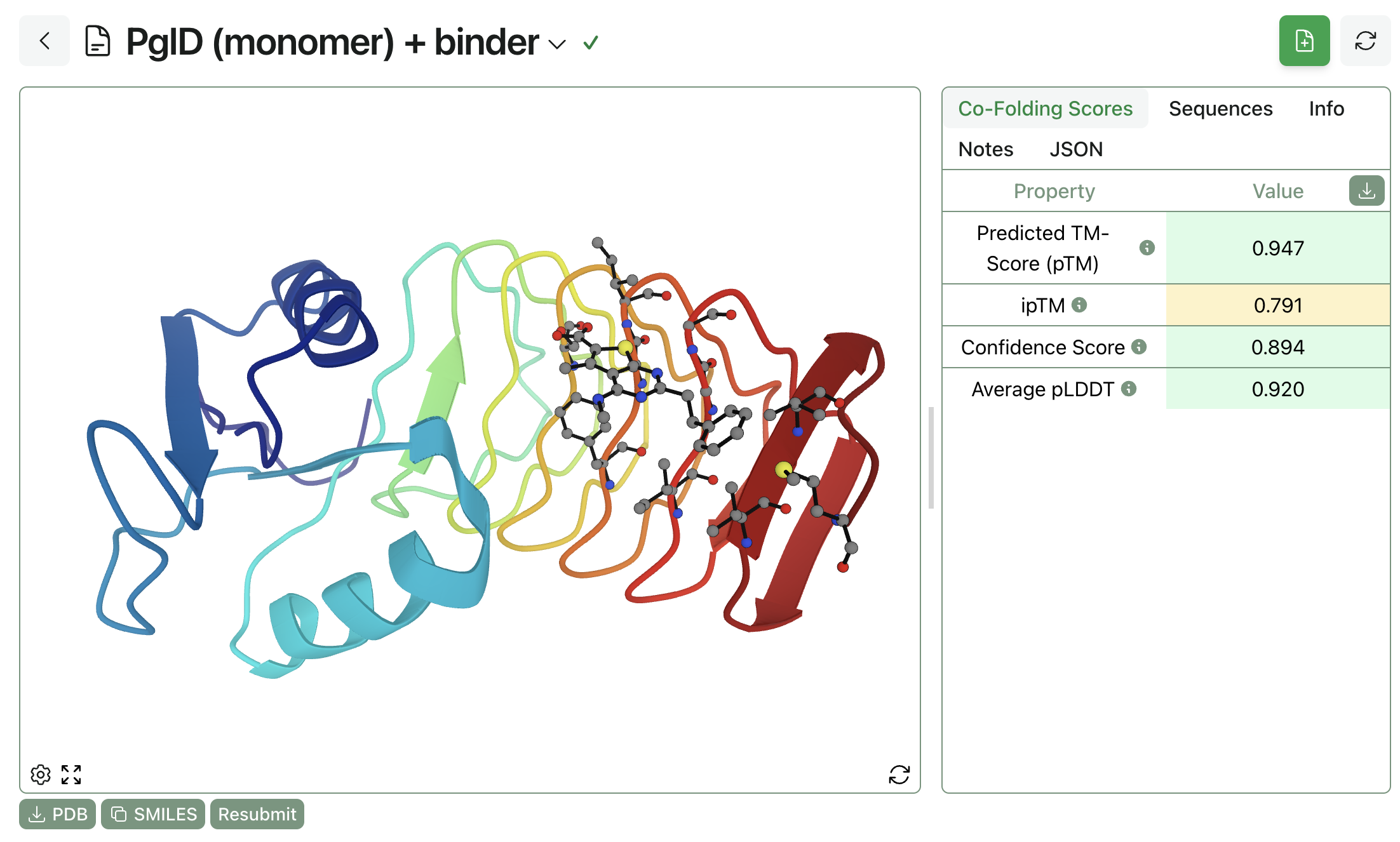
Python API Access
Boltz-1 calculations can also easily be submitted via Rowan's Python API, which is available for free to all users:
import rowan
rowan.api_key = "your-rowan-api-key"
workflow = rowan.submit_protein_cofolding_workflow(
initial_protein_sequences=[
"MENFQKVEKIGEGTYGVVYKARNKLTGEVVALKKIRLDTETEGVPSTAIREISLLKELNHPNIVKLLDVIHTENKLYLVFEFLHQDLKKFMDASALTGIPLPLIKSYLFQLLQGLAFCHSHRVLHRDLKPQNLLINTEGAIKLADFGLARAFGVPVRTYTHEVVTLWYRAPEILLGCKYYSTAVDIWSLGCIFAEMVTRRALFPGDSEIDQLFRIFRTLGTPDEVVWPGVTSMPDYKPSFPKWARQDFSKVVPPLDEDGRSLLSQMLHYDPNKRISAKAALAHPFFQDVTKPVPHLRL"
],
initial_smiles_list=["CCC(C)CN=C1NCC2(CCCOC2)CN1"],
name="Boltz-1 cofolding job",
model="boltz_1",
)
workflow.wait_for_result()
workflow.fetch_latest(in_place=True)
print(workflow.data)
print(f"View the 3D structure on the web at labs.rowansci.com/protein-cofolding/{workflow.uuid}")
This prints the following data; the corresponding 3D structure can be viewed through Rowan's web interface.
{
'lddt': [0.839, 0.911, 0.942, 0.977, 0.95, 0.958, 0.968, 0.965, 0.968, 0.895, 0.903, 0.898, 0.848, 0.868, 0.891, 0.904, 0.936, 0.956, 0.977, 0.983, 0.984, 0.982, 0.97, 0.969, 0.914, 0.941, 0.973, 0.946, 0.98, 0.988, 0.978, 0.978, 0.961, 0.944, 0.939, 0.757, 0.642, 0.58, 0.496, 0.558, 0.584, 0.714, 0.821, 0.882, 0.903, 0.919, 0.939, 0.944, 0.948, 0.958, 0.951, 0.959, 0.948, 0.958, 0.97, 0.97, 0.967, 0.964, 0.974, 0.988, 0.988, 0.99, 0.985, 0.985, 0.984, 0.982, 0.98, 0.973, 0.97, 0.967, 0.949, 0.919, 0.883, 0.889, 0.908, 0.957, 0.972, 0.976, 0.98, 0.979, 0.966, 0.98, 0.982, 0.94, 0.974, 0.971, 0.989, 0.976, 0.974, 0.989, 0.989, 0.985, 0.983, 0.986, 0.977, 0.975, 0.964, 0.986, 0.989, 0.987, 0.989, 0.989, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.989, 0.989, 0.99, 0.988, 0.982, 0.988, 0.987, 0.978, 0.958, 0.979, 0.97, 0.981, 0.958, 0.956, 0.985, 0.974, 0.987, 0.955, 0.885, 0.989, 0.983, 0.989, 0.987, 0.985, 0.982, 0.989, 0.989, 0.99, 0.989, 0.987, 0.971, 0.923, 0.876, 0.887, 0.91, 0.902, 0.921, 0.925, 0.896, 0.817, 0.719, 0.817, 0.817, 0.88, 0.834, 0.788, 0.782, 0.837, 0.83, 0.868, 0.898, 0.949, 0.98, 0.988, 0.988, 0.98, 0.989, 0.99, 0.989, 0.981, 0.988, 0.99, 0.987, 0.983, 0.953, 0.965, 0.953, 0.986, 0.987, 0.99, 0.987, 0.989, 0.99, 0.99, 0.989, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.989, 0.988, 0.984, 0.989, 0.99, 0.99, 0.989, 0.988, 0.974, 0.98, 0.981, 0.989, 0.989, 0.989, 0.989, 0.99, 0.99, 0.99, 0.99, 0.989, 0.99, 0.99, 0.99, 0.99, 0.99, 0.989, 0.99, 0.988, 0.989, 0.99, 0.989, 0.989, 0.99, 0.99, 0.987, 0.988, 0.988, 0.989, 0.99, 0.989, 0.989, 0.989, 0.99, 0.99, 0.989, 0.99, 0.988, 0.988, 0.982, 0.935, 0.94, 0.979, 0.982, 0.987, 0.988, 0.976, 0.932, 0.989, 0.987, 0.987, 0.989, 0.99, 0.988, 0.989, 0.99, 0.99, 0.986, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.989, 0.988, 0.987, 0.99, 0.99, 0.99, 0.99, 0.989, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.989, 0.989, 0.99, 0.989, 0.989, 0.988, 0.973, 0.987, 0.949, 0.974, 0.916, 0.946, 0.477, 0.542, 0.639, 0.543, 0.629, 0.749, 0.815, 0.806, 0.775, 0.746, 0.584, 0.521, 0.527, 0.535, 0.613, 0.714, 0.798],
'model': 'boltz_1',
'scores': {'ptm': 0.964, 'iptm': 0.747, 'avg_lddt': 0.941, 'confidence_score': 0.902},
'messages': [],
'affinity_score': None,
'use_msa_server': True,
'use_potentials': False,
'pocket_constraints': [],
'contact_constraints': [],
'initial_smiles_list': ['CCC(C)CN=C1NCC2(CCCOC2)CN1'],
'use_templates_server': False,
'predicted_structure_uuid': '1be2e3d4-eb17-4e78-8ec0-2cd88bb7c2a4',
'initial_protein_sequences': ['MENFQKVEKIGEGTYGVVYKARNKLTGEVVALKKIRLDTETEGVPSTAIREISLLKELNHPNIVKLLDVIHTENKLYLVFEFLHQDLKKFMDASALTGIPLPLIKSYLFQLLQGLAFCHSHRVLHRDLKPQNLLINTEGAIKLADFGLARAFGVPVRTYTHEVVTLWYRAPEILLGCKYYSTAVDIWSLGCIFAEMVTRRALFPGDSEIDQLFRIFRTLGTPDEVVWPGVTSMPDYKPSFPKWARQDFSKVVPPLDEDGRSLLSQMLHYDPNKRISAKAALAHPFFQDVTKPVPHLRL'],
'ligand_binding_affinity_index': None
}
If you're interested in trying out Boltz-1 today, make an account for free and start running calculations in minutes!
