Multi-Modal Stucture Prediction with Chai-1
For a comparison against Rowan's other co-folding models, see our co-folding overview page.
Chai-1 is a general-purpose deep learning model for biomolecular structure prediction, designed to work across a wide range of inputs: proteins, nucleic acids, small-molecule ligands, and multi-chain assemblies. Unlike models specialized for a single domain, Chai-1 supports multiple modalities in one architecture and can be optionally guided by experimental constraints such as cross-linking mass spectrometry, epitope mapping, or known contact residues.
Because Chai-1 can run in both full multi-sequence-alignment (MSA) mode and single-sequence mode, it adapts well to different stages of discovery—from high-throughput screening against well-characterized targets to exploratory modeling of novel or variable sequences where evolutionary data is sparse.
Method Overview
Chai-1 follows a paired-attention architecture similar in spirit to AlphaFold3, but with key differences. Chai-1 can incorporate residue-level embeddings from protein language models, enabling strong single-sequence performance. Chai-1 can also incorporate optional pocket, contact, and docking restraints to bias the model's predictions in the direction of experimental priors. The full architecture diagram is shown here:
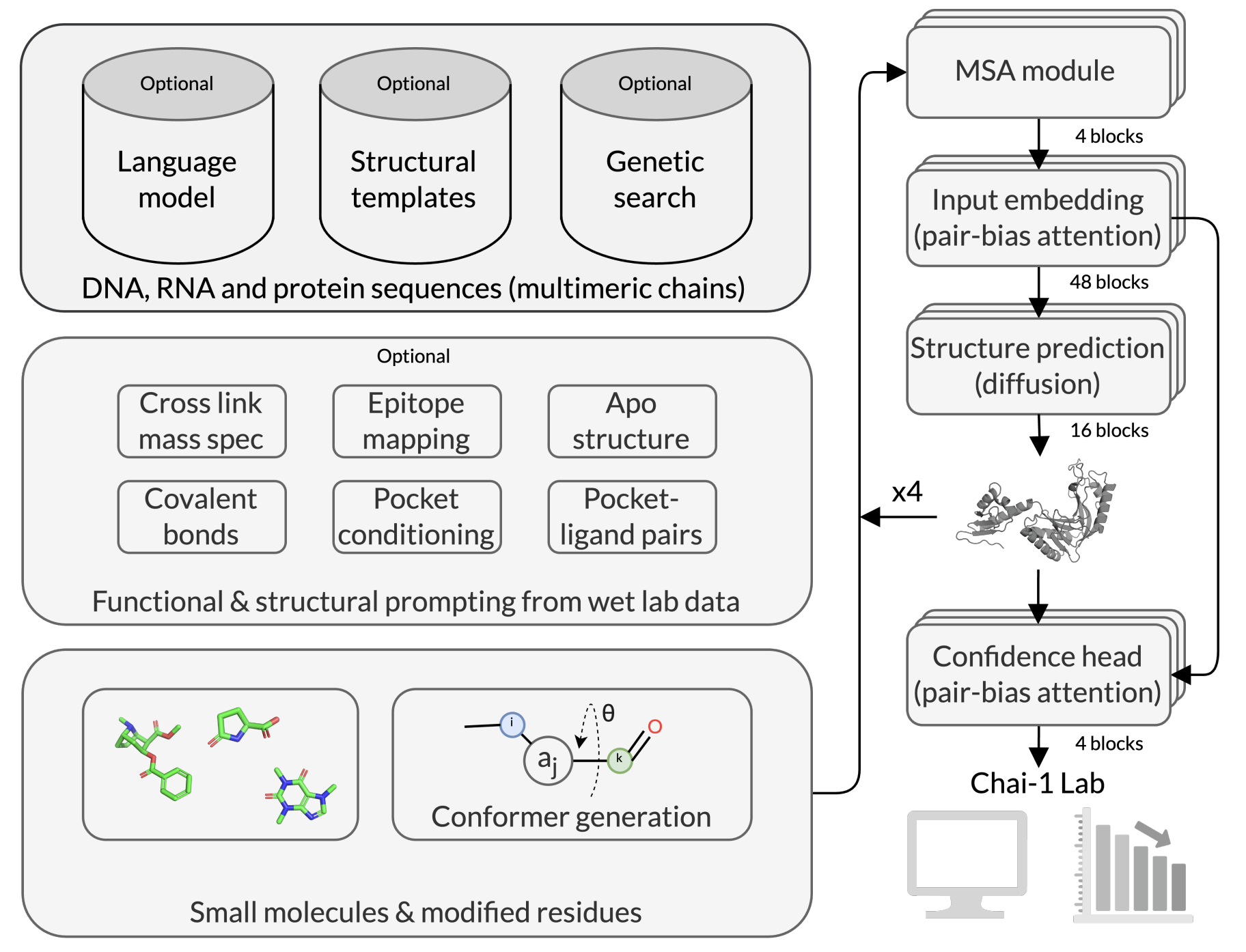
The architecture of Chai-1 (Figure 1 from the preprint).
During inference, Chai-1 can sample multiple structures (via trunk and diffusion sampling) and ranks them by calibrated confidence metrics such as interface pTM (ipTM) (Figure 2 from the preprint). This is only a brief summary of how Chai-1 works; for full details, see the preprint.
Performance
On the PoseBusters protein–ligand benchmark, restricted to post-cutoff structures, Chai-1 achieves a 77% success rate (ligand RMSD ≤ 2 Å) from sequence + SMILES alone, comparable to AlphaFold3's reported 76%. When provided the apo protein structure as a prompt, success rises to 81%.
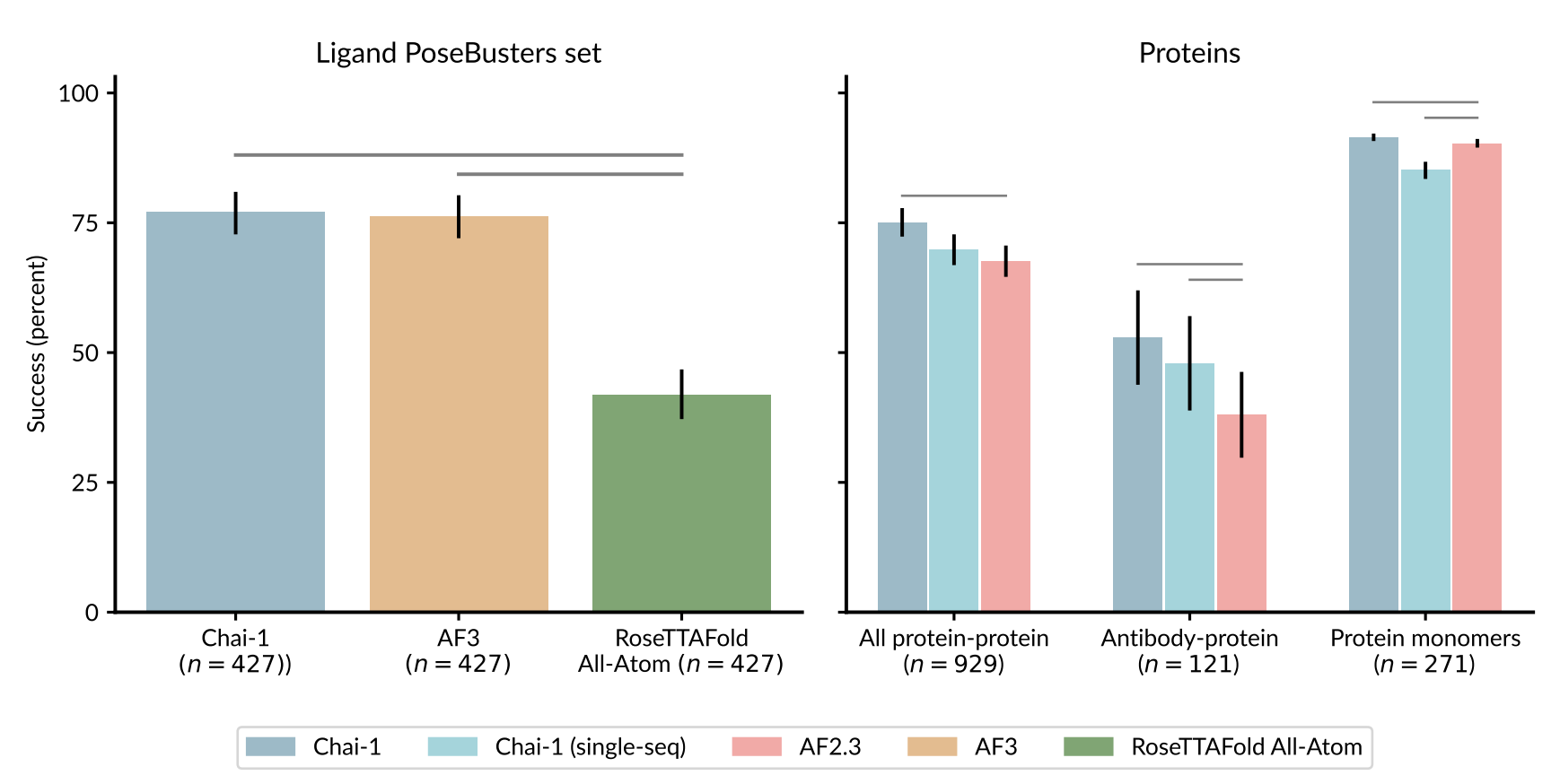
Protein–ligand pose prediction success rates (PoseBusters V1) and protein accuracy (DockQ and RMSD). See the preprint for full details.
For protein–protein complexes in a low-homology set, Chai-1 with MSAs outperforms AlphaFold-Multimer 2.3 (0.751 vs 0.677 DockQ success). In single-sequence mode without MSAs, it still matches or exceeds AF2.3 on many interface types, particularly antibody–protein complexes, where evolutionary signal is often weak.
Chai-1 also supports nucleic acid modeling without MSAs, achieving accuracy comparable to RoseTTAFold2NA despite lacking evolutionary inputs for RNA/DNA.
Nevertheless, a word of caution is warranted. Benchmarks from Peter Škrinjar and co-workers demonstrate that the performance of current protein–ligand co-folding methods, including Chai-1, depends largely on similarity to the training set. This suggests that particular caution is warranted for novel or poorly explored regions of biochemical space.
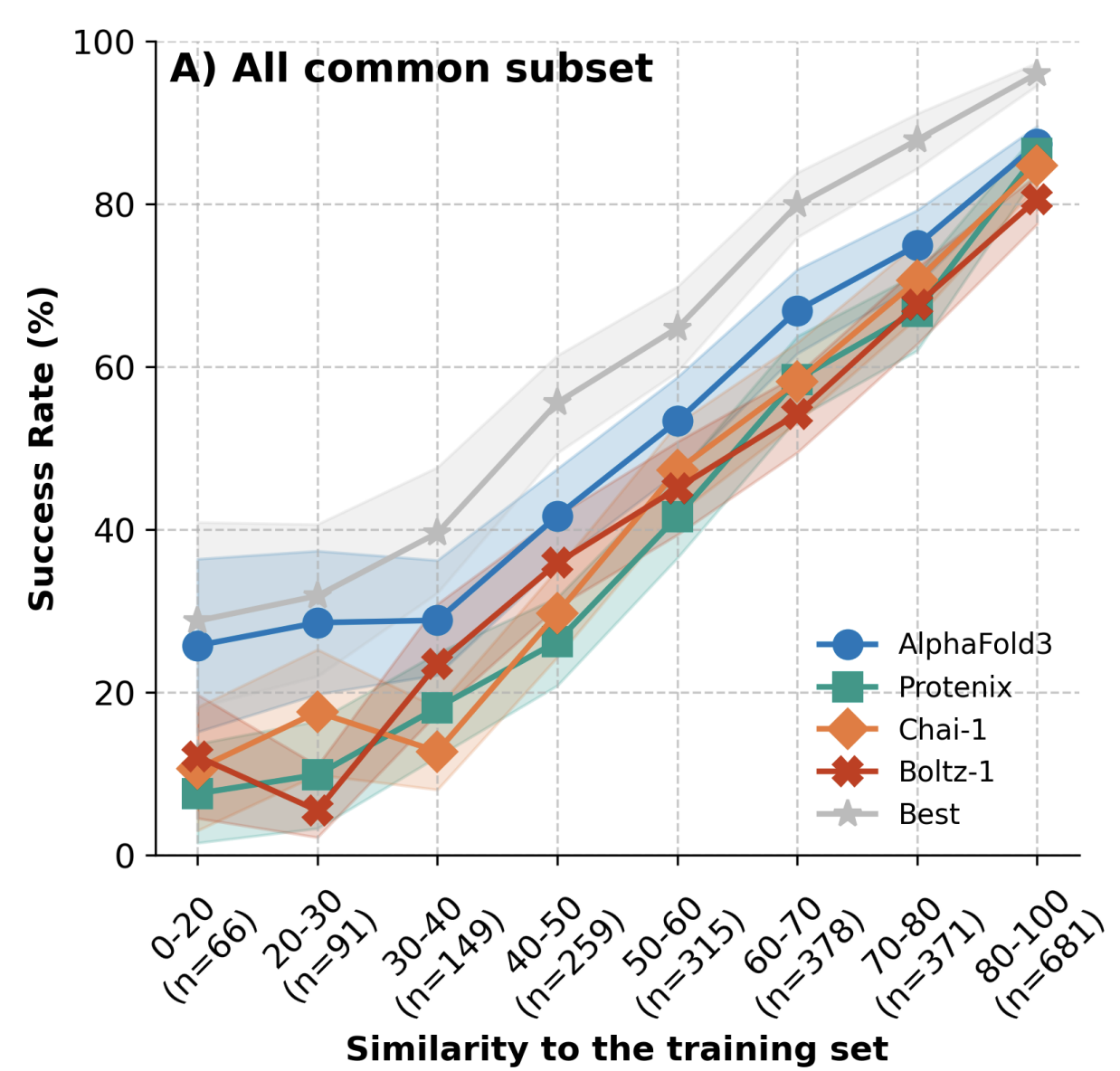
Similarity to the training set predicts cofolding performance.
(On Practical Cheminformatics, Pat Walters discusses this paper and a variety of other studies demonstrating that Chai-1 and related methods don't yet understand binding physics correctly.)
Applications
Chai-1's breadth and ability to run without MSAs makes it suitable for ligand pose generation, protein–protein docking, antibody–antigen modeling, nucleic-acid-complex prediction without the need for specialized MSAs, or simply exploratory protein-structure modeling.
How does this work in practice? Here's what Pat Walters has to say about the current use of co-folding methods in drug discovery:
I have been using co-folding methods in a similar way to how I use molecule generation techniques— as tools to develop hypotheses that can be tested experimentally. We have many methods that tell us if a molecule binds, such as biochemical assays, size exclusion chromatography-MS, examining binding in a DEL screen, or performing various other experiments. However, apart from X-ray crystallography or cryoEM, we have very few experiments that can definitively identify where a compound binds. In most cases, we need a hypothesis to guide experiments, such as mutagenesis or installing a photoaffinity label. Even more definitive experiments, like NMR or HDX-MS, are much more useful when a binding hypothesis is available. Co-folding provides an easy way to generate these hypotheses; then, it's up to our scientific knowledge, intuition, and creativity to decide how to proceed.
This conclusion—that co-folding methods are promising hypothesis generators but no substitute for experimental data—seems to match what we're seeing from lots of other drug-discovery teams.
Accessing Chai-1 in Rowan
Rowan provides browser-based access to Chai-1, with support for all major input types. Users can run Chai-1 in:
- Full mode (with MSA search) for maximum accuracy.
- Single-sequence mode for faster turnaround or novel sequences.
An interactive 3D viewer lets you inspect predicted contacts, ligand geometry, and inter-chain interfaces.
Here's what the result of a Chai-1 calculation looks like on Rowan:
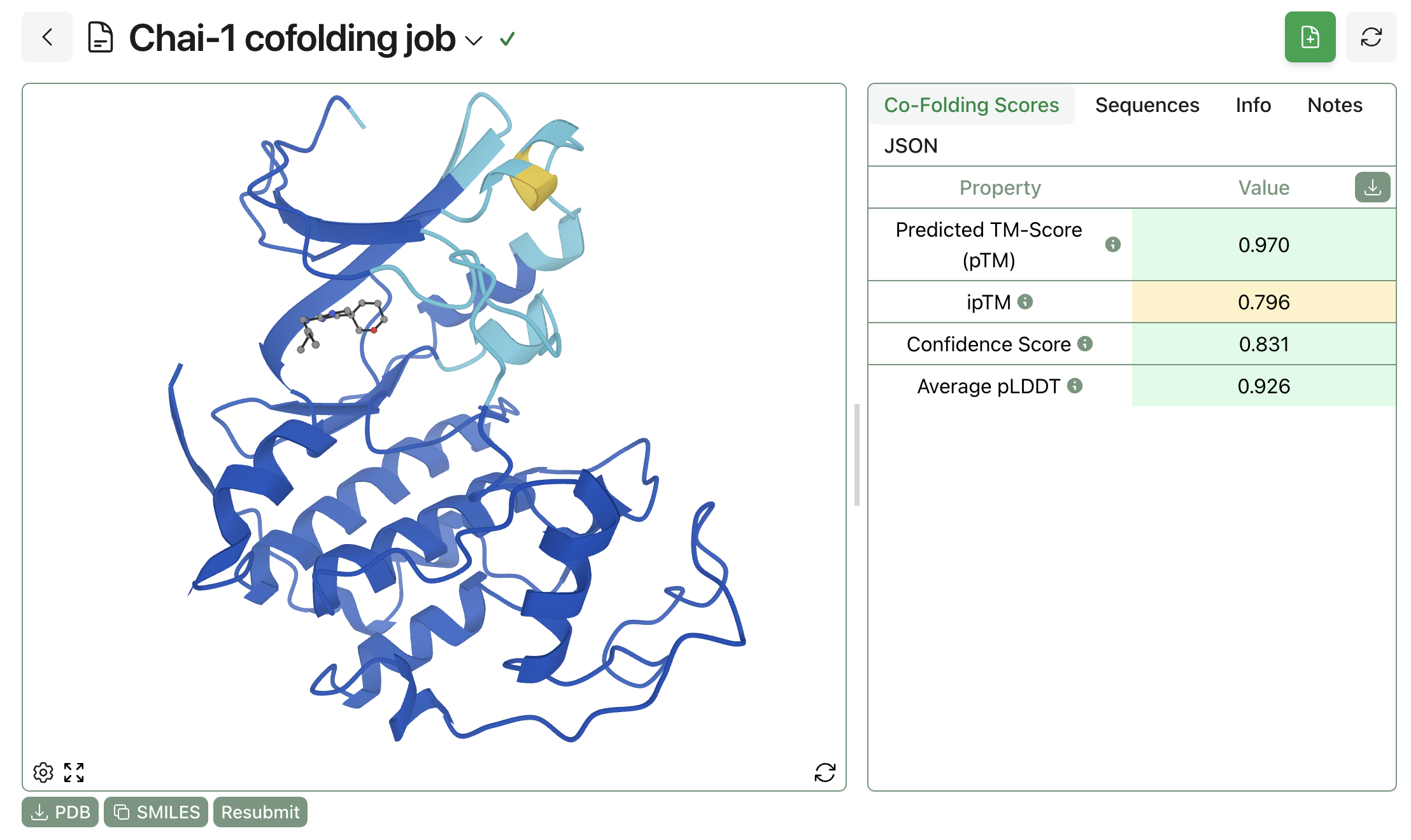
Python API Access
Chai-1 calculations can also easily be submitted via Rowan's Python API, which is available for free to all users:
import rowan
rowan.api_key = "your-rowan-api-key"
workflow = rowan.submit_protein_cofolding_workflow(
initial_protein_sequences=[
"MENFQKVEKIGEGTYGVVYKARNKLTGEVVALKKIRLDTETEGVPSTAIREISLLKELNHPNIVKLLDVIHTENKLYLVFEFLHQDLKKFMDASALTGIPLPLIKSYLFQLLQGLAFCHSHRVLHRDLKPQNLLINTEGAIKLADFGLARAFGVPVRTYTHEVVTLWYRAPEILLGCKYYSTAVDIWSLGCIFAEMVTRRALFPGDSEIDQLFRIFRTLGTPDEVVWPGVTSMPDYKPSFPKWARQDFSKVVPPLDEDGRSLLSQMLHYDPNKRISAKAALAHPFFQDVTKPVPHLRL"
],
initial_smiles_list=["CCC(C)CN=C1NCC2(CCCOC2)CN1"],
name="Chai-1 cofolding job",
model="chai_1r",
)
workflow.wait_for_result()
workflow.fetch_latest(in_place=True)
print(workflow.data)
This prints the following data; the corresponding 3D structure can be viewed through Rowan's web interface.
{
'lddt': [0.895, 0.938, 0.967, 0.976, 0.971, 0.971, 0.971, 0.97, 0.963, 0.94, 0.908, 0.873, 0.835, 0.838, 0.836, 0.874, 0.921, 0.945, 0.968, 0.977, 0.982, 0.981, 0.977, 0.97, 0.948, 0.961, 0.968, 0.973, 0.982, 0.982, 0.976, 0.964, 0.941, 0.921, 0.889, 0.771, 0.72, 0.691, 0.643, 0.678, 0.716, 0.742, 0.784, 0.832, 0.846, 0.881, 0.885, 0.892, 0.895, 0.917, 0.909, 0.931, 0.924, 0.907, 0.937, 0.942, 0.94, 0.95, 0.967, 0.984, 0.987, 0.988, 0.984, 0.981, 0.973, 0.967, 0.968, 0.958, 0.951, 0.938, 0.89, 0.862, 0.796, 0.735, 0.837, 0.902, 0.942, 0.953, 0.969, 0.974, 0.978, 0.982, 0.98, 0.965, 0.98, 0.982, 0.988, 0.982, 0.98, 0.987, 0.987, 0.984, 0.981, 0.982, 0.977, 0.974, 0.969, 0.984, 0.987, 0.987, 0.987, 0.989, 0.989, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.989, 0.99, 0.989, 0.986, 0.986, 0.987, 0.986, 0.975, 0.978, 0.978, 0.967, 0.951, 0.956, 0.964, 0.957, 0.942, 0.946, 0.969, 0.973, 0.983, 0.96, 0.961, 0.986, 0.985, 0.987, 0.984, 0.984, 0.983, 0.988, 0.989, 0.989, 0.988, 0.985, 0.972, 0.916, 0.881, 0.83, 0.86, 0.866, 0.869, 0.84, 0.826, 0.778, 0.791, 0.778, 0.796, 0.845, 0.804, 0.759, 0.784, 0.811, 0.835, 0.843, 0.859, 0.925, 0.969, 0.983, 0.981, 0.98, 0.985, 0.988, 0.987, 0.979, 0.983, 0.987, 0.982, 0.97, 0.939, 0.949, 0.909, 0.978, 0.983, 0.987, 0.983, 0.984, 0.989, 0.989, 0.987, 0.99, 0.99, 0.99, 0.989, 0.99, 0.99, 0.989, 0.989, 0.989, 0.988, 0.987, 0.986, 0.988, 0.989, 0.989, 0.986, 0.979, 0.958, 0.968, 0.969, 0.986, 0.987, 0.987, 0.989, 0.99, 0.989, 0.99, 0.99, 0.989, 0.989, 0.989, 0.989, 0.99, 0.989, 0.989, 0.989, 0.988, 0.988, 0.989, 0.988, 0.987, 0.988, 0.989, 0.987, 0.987, 0.987, 0.988, 0.989, 0.988, 0.988, 0.988, 0.989, 0.989, 0.989, 0.989, 0.988, 0.987, 0.985, 0.964, 0.97, 0.985, 0.985, 0.985, 0.988, 0.978, 0.966, 0.986, 0.986, 0.987, 0.989, 0.989, 0.989, 0.989, 0.99, 0.989, 0.988, 0.99, 0.99, 0.99, 0.989, 0.99, 0.989, 0.989, 0.987, 0.988, 0.989, 0.99, 0.989, 0.99, 0.989, 0.99, 0.99, 0.99, 0.989, 0.99, 0.989, 0.99, 0.99, 0.988, 0.988, 0.989, 0.989, 0.989, 0.989, 0.985, 0.985, 0.971, 0.971, 0.939, 0.942],
'model': 'chai_1r',
'scores': {'ptm': 0.97, 'iptm': 0.796, 'avg_lddt': 0.926, 'confidence_score': 0.831},
'messages': [],
'affinity_score': None,
'use_msa_server': True,
'use_potentials': False,
'pocket_constraints': [],
'contact_constraints': [],
'initial_smiles_list': ['CCC(C)CN=C1NCC2(CCCOC2)CN1'],
'use_templates_server': False,
'predicted_structure_uuid': '0d3bc244-e7f2-473d-bd4a-439b1e03a2e8',
'initial_protein_sequences': ['MENFQKVEKIGEGTYGVVYKARNKLTGEVVALKKIRLDTETEGVPSTAIREISLLKELNHPNIVKLLDVIHTENKLYLVFEFLHQDLKKFMDASALTGIPLPLIKSYLFQLLQGLAFCHSHRVLHRDLKPQNLLINTEGAIKLADFGLARAFGVPVRTYTHEVVTLWYRAPEILLGCKYYSTAVDIWSLGCIFAEMVTRRALFPGDSEIDQLFRIFRTLGTPDEVVWPGVTSMPDYKPSFPKWARQDFSKVVPPLDEDGRSLLSQMLHYDPNKRISAKAALAHPFFQDVTKPVPHLRL'],
'ligand_binding_affinity_index': None
}
If you're interested in trying out Chai-1 today, make an account for free and start running calculations in minutes!
