Hydrogen-Bond Basicity
Hydrogen bonding is a fundamental non-covalent interactions, and hydrogen bonds are often key components of biological systems or small-molecule drugs. Tuning the strength of hydrogen-bond donors and acceptors can be used to control a litany of properties important in drug design, like P-glycoprotein transport, efflux ratio, lipophilicity, and blood–brain barrier permeability.
Quantifying Hydrogen-Bond-Acceptor Strength
Hydrogen-bond-acceptor strength can be quantified experimentally as pKBHX, the base-10 logarithm of the association constant with a model hydrogen-bond donor. For historical and practical reasons, 4-fluorophenol is typically used as the hydrogen-bond donor and carbon tetrachloride is typically used as the solvent; values measured under these conditions correlate well with other choices of hydrogen-bond donor and solvent.
Under these conditions, neutral hydrogen-bond acceptors typically have pKBHX values between about −1 and 5. Here's a plot of typical pKBHX values by functional group:
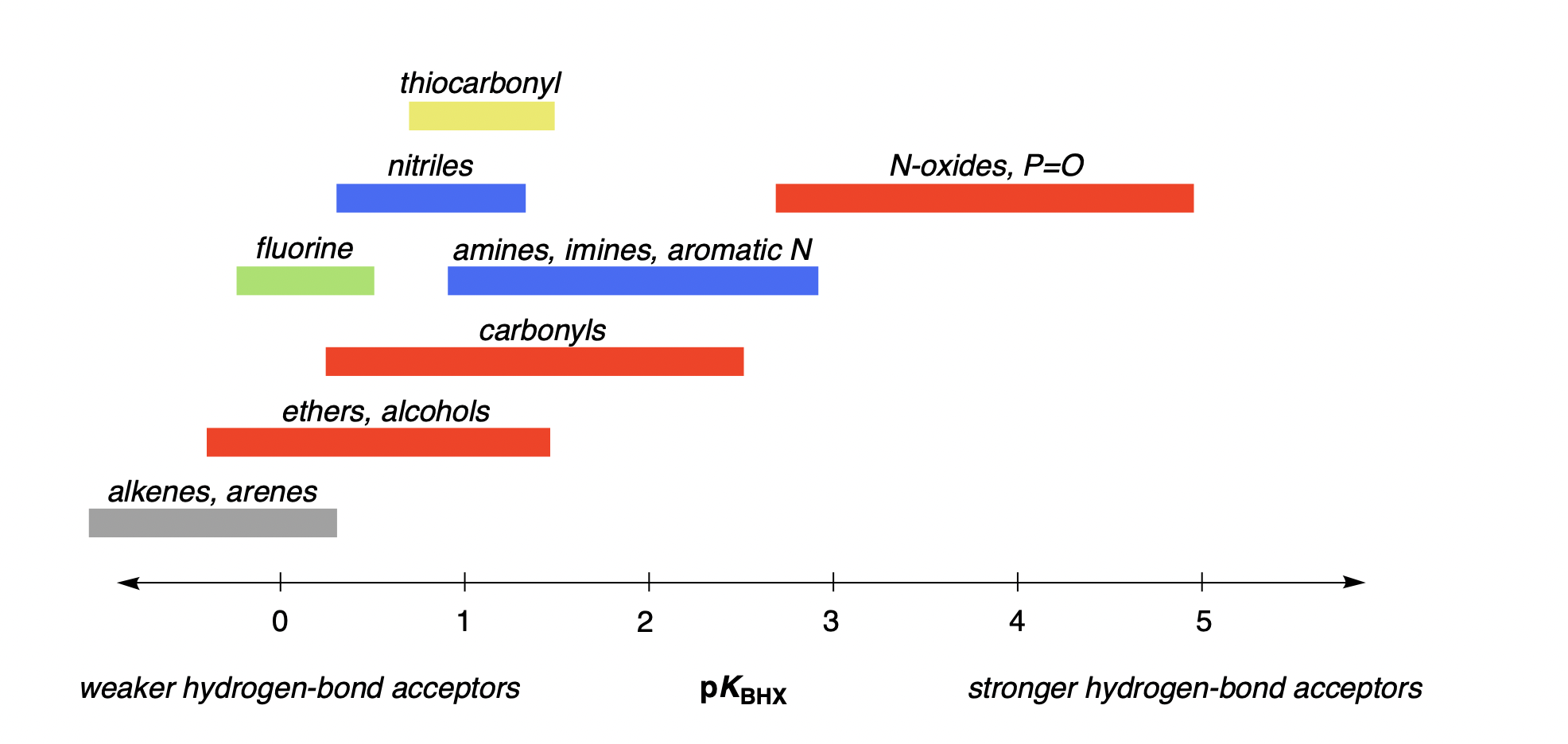
Predicting Hydrogen-Bond-Basicity Values Computationally
As discussed above, pKBHX values can be determined experimentally by measuring association constants in the lab (with spectroscopy or similar techniques). However, these pKBHX predictions are not typically broken down by hydrogen-bond-acceptor site, making them of limited utility for complex molecules. Furthermore, accurate measurement of association constants is time-consuming and requires sufficient purified material, making it poorly suited for exploratory screening or ideation.
For this reason, predicting hydrogen-bond-acceptor strength in silico has been a goal of computational chemists for decades. Since the 1990s, researchers have found that the minimum electrostatic potential Vmin in the region of lone pairs can be used to predict per-site pKBHX values via linear regression, with different slopes and intercepts for each functional group. Despite this advance, these workflows are difficult to run and still not commonly employed.
Rowan's Hydrogen-Bond-Basicity Workflow
At Rowan, we've recently developed a streamlined algorithm for predicting the hydrogen-bond basicity of arbitrary drug-like molecules. Our approach uses neural network potentials to accelerate conformer optimization and screening, and uses only a single low-cost r2SCAN-3c calculation per molecule studied, allowing predictions to be made in minutes, not days. We've released our workflow as a preprint.
Here's the result of a hydrogen-bond-basicity prediction visualized through Rowan:
Real-World Impact
Rowan's hydrogen-bond-basicity-prediction workflow has already been used to interpret real-world trends in complex drug-discovery projects. In a recent LinkedIn post, Gilles Ouvry used Rowan's pKBHX predictions to understand a "magic chloro" effect observed by Genentech in pyrazole-containing LRRK2 inhibitors. Switching from methyl to chloro not only increased the half-life of the compound by eliminating a metabolic hotspot but also decreased efflux and improved Kp,uu (blood–brain-barrier penetrance). Ouvry showed that these trends could be nicely explained by showing that chloro substitution made the pyrazole nitrogen a weaker hydrogen-bond acceptor, which presumably decreases transporter recognition.
