Molecular Orbitals, Isosurfaces, and More
Molecular orbitals describe the location and density of electrons in a molecule. They can be sensitive indicators of molecular properties, such as nucleo/electrophilicity, UV-stability, redox potential, and much more. Molecular orbitals are formed from combinations of atomic orbitals and come in bonding, non-bonding, and anti-bonding forms. Orbitals can be spread across a whole molecule or localized to individual atoms or fragments.
Types of Molecular Orbitals
Frontier orbitals (i.e. the highest energy occupied orbitals and lowest energy unoccupied orbitals) contribute the most to chemical reactivity and photophysical properties. The location of frontier orbitals can indicate where a reaction will take place, while the energy levels indicate the relative willingness to accept or donate an electron. The Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) are indicators of the reduction potential and oxidation potential. The gap between them (often referred to as the HOMO–LUMO gap) can be a sensitive indicator of the UV-stability of a molecule, and modifications to the molecule to increase this gap can lead to significant increases in lifespan of the molecule. The overlap of orbitals between molecules can predict the flow of electrons and holes in OLEDs and other electronics, allowing one to finely tune the efficiency of this transfer.
Core orbitals have less of an effect on reactivity, but they are fundamental to any high-energy spectroscopy techniques. X-ray absorption spectroscopy measures the energy of the core electrons by ejecting them with high-energy x-rays, giving a sensitive indicator of the elements in a material. Bonding information can be derived from the fine structure of the resulting spectra via x-ray near edge spectroscopy (XANES) and x-ray absorption fine structure (EXAFS), with the core and frontier orbital energies being fundamental to understanding the results. Computing the energies of these core orbitals and how orbitals are perturbed when bonded to other elements can help identify materials, the arrangement of atoms in a molecule, and the oxidation state of metals.
Plotting Molecular Orbitals
Rowan's orbitals workflow can compute any molecular orbital for a given molecule, occupied or unoccupied. This is a visualization of the lowest energy π-type bonding orbitals of benzene, illustrating two regions of electron density above and below the ring:
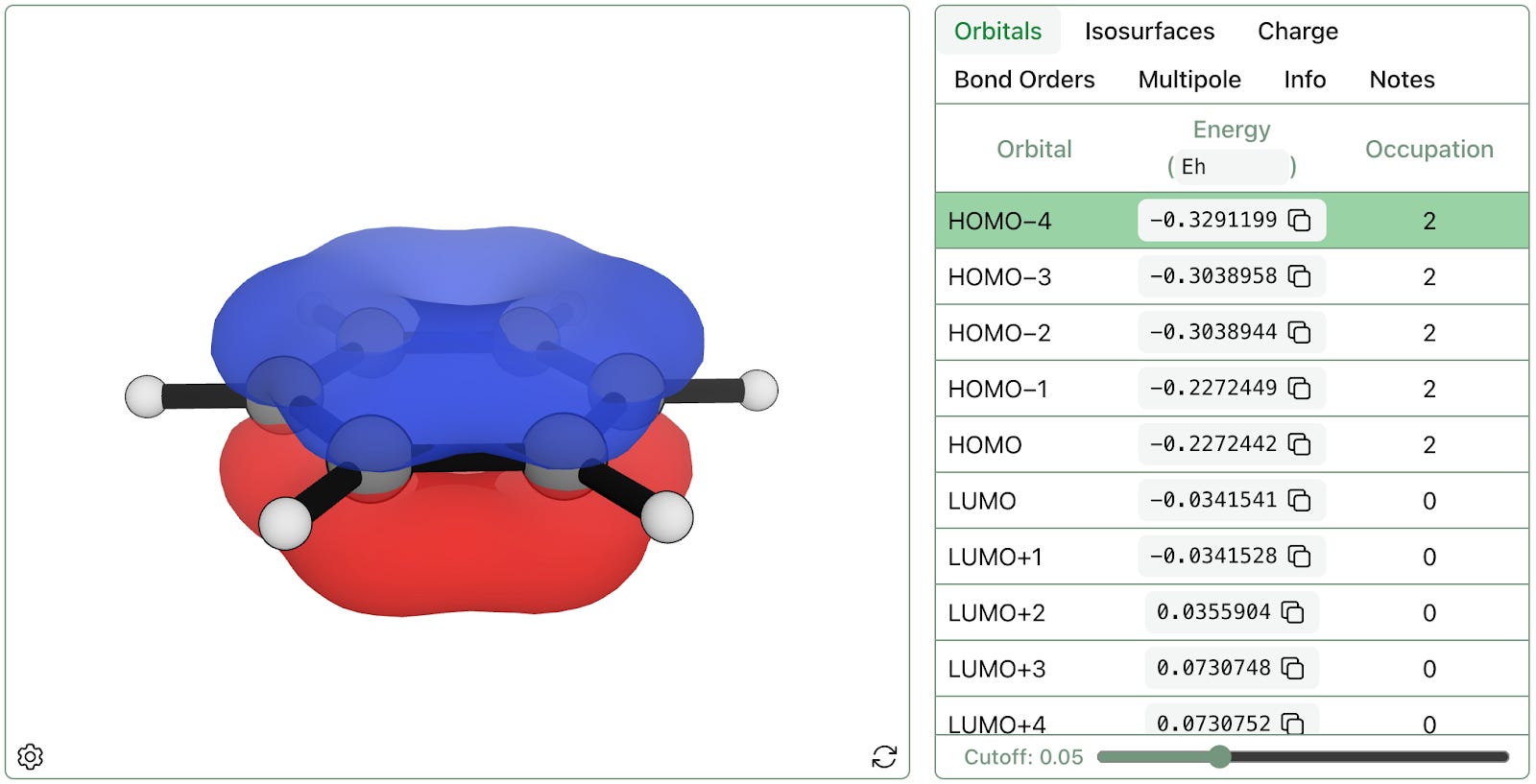
The HOMO-4 bonding orbital of benzene.
Isosurfaces
Rowan's orbitals workflow also computes the molecular electrostatic potential, electron density, and spin density. Rowan uses 3D isosurfaces to visualize these properties, making it simple to see where electrons reside in space and estimate molecular volume. Here's an illustration of a density isosurface of water, colored by electrostatic potential:
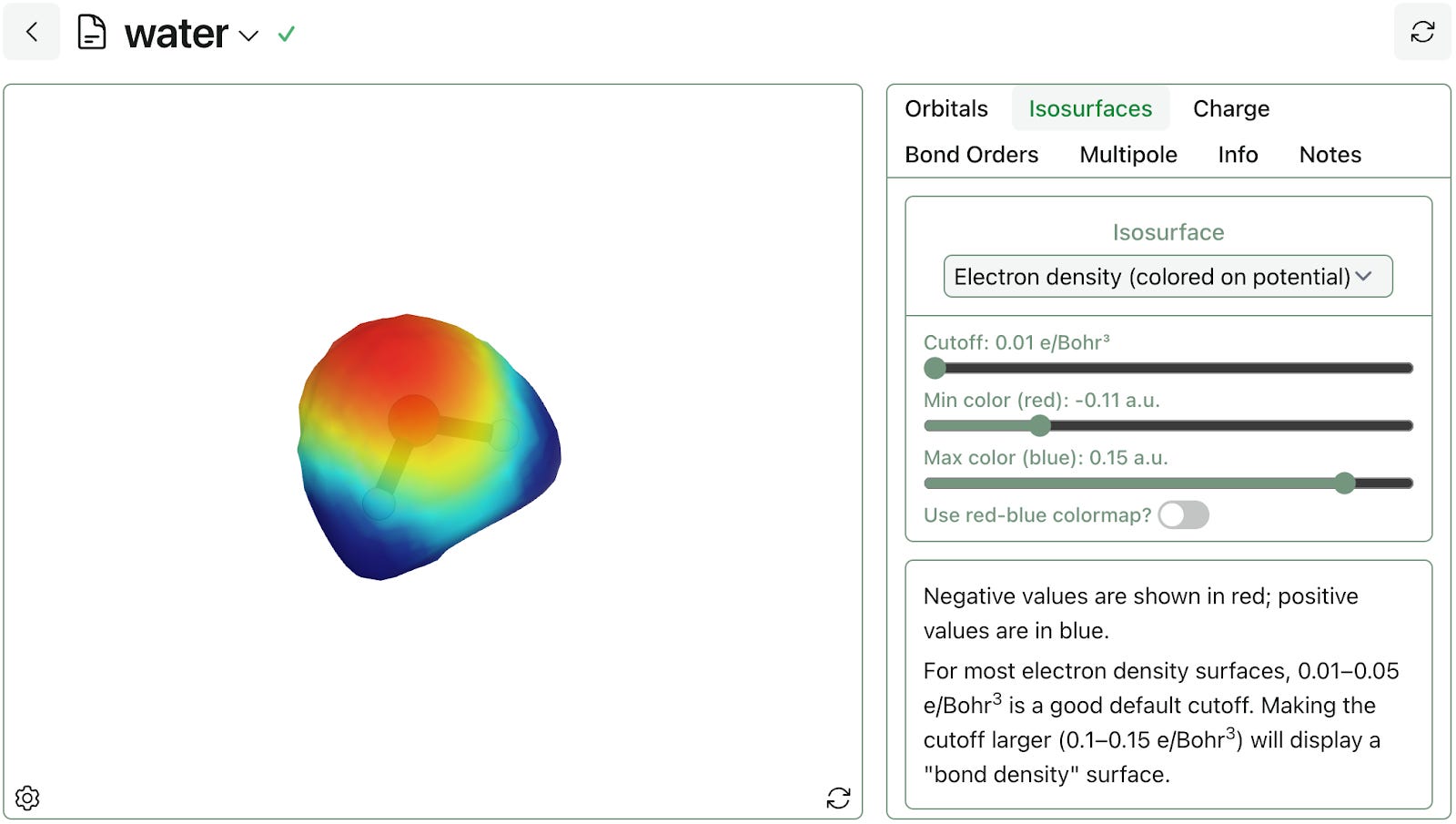
The oxygen has partial negative charge, while the hydrogens have partial positive charge.
Rowan allows users to adjust the cutoff used for isosurface visualization. With cutoffs in the range of 0.01–0.05 e/Bohr3), density isosurfaces can provide a sense of a molecule's shape. With a higher cutoff (0.1–0.15 e/Bohr3), the same isosurfaces can be used to visualize "bond density" (areas with higher order bonds will have correspondingly higher isosurface volumes).
The electrostatic potential isosurface allows scientists to visualize a molecule's charge distribution. Areas of negative electrostatic potential correspond to areas that will attract positive charges (electrophiles), while areas of positive electrostatic potential correspond to areas that will attract negative charges (nucleophiles). These visualizations can be used to understand the origin of attractive or repulsive non-covalent interactions, like the σ-holes implicated in halogen and chalogen bonding.
For open-shell systems, the spin density isosurface can also be visualized to illustrate where unpaired electrons are located. Viewing a spin density isosurface for TEMPO shows that the unpaired electrons live in the N–O bond, as expected:
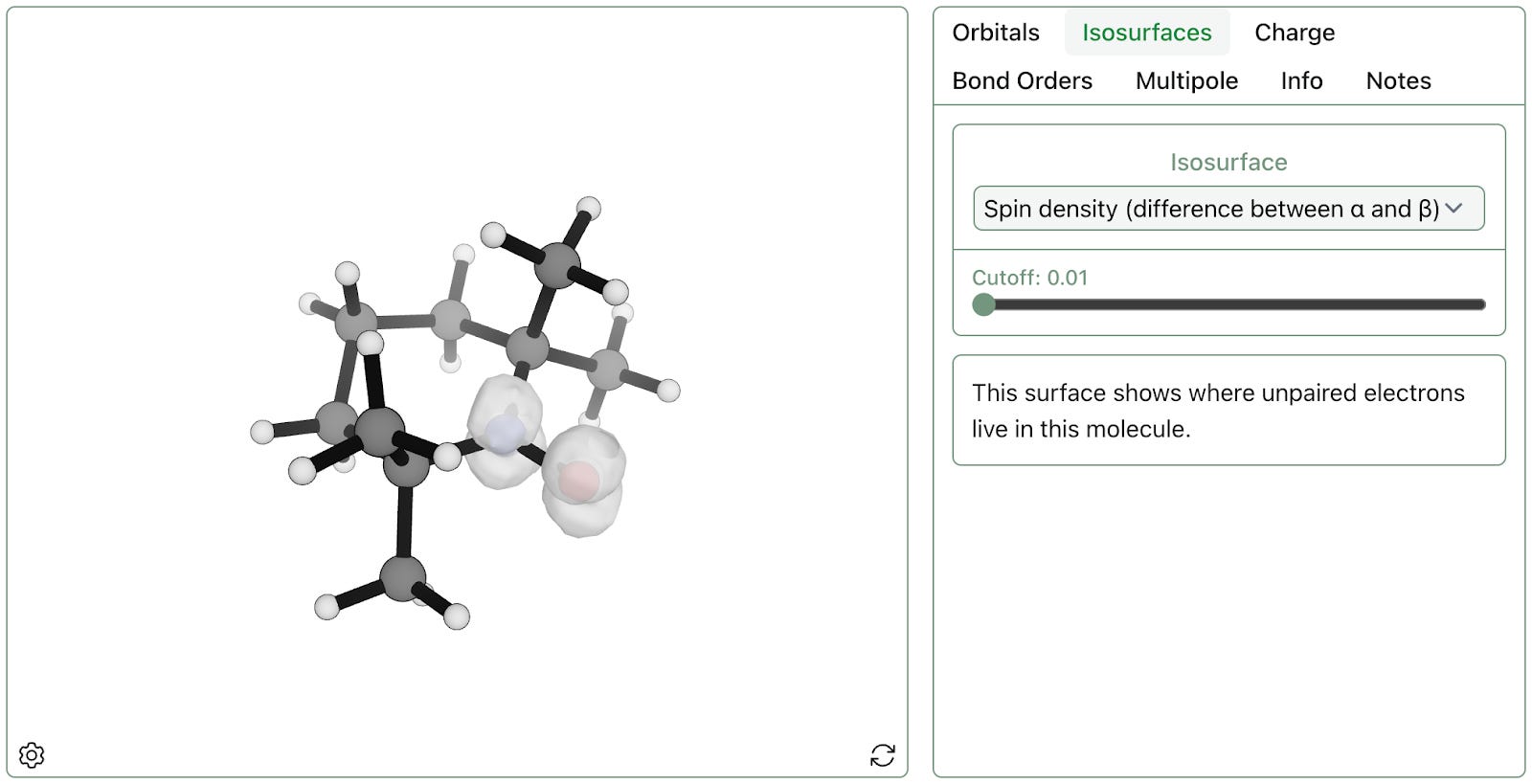
As expected, the N–O moiety contains the radical.
Bond Order, Charges, and Multipole Moments
Computing orbitals through Rowan also computes Wiberg bond order, Mayer bond order, Löwdin charges, Mulliken charges, the molecular dipole moment, and the molecular quadrupole moment. These values all provide complementary information on where electrons reside. Bond order can be useful in gaining insight into aromaticity and bond localization and delocalization, while charges and multipole moments can be used to determine where electron density resides and can be used as descriptors in QSAR/QSPR models or to parameterize molecular force fields.
Rowan's orbital workflow provides scientists with a wide range of information about electronic structure and molecular properties, with intuitive publication-ready visualizations available right there in the web browser.
