Computing Conformational Strain
When presented with an unfamiliar molecular conformation, scientists often wonder how strained that conformation is relative to the molecule's lowest-energy conformation. This question arises across various subfields of chemistry: strain computations can be key to understanding substrate–catalyst interactions, and computing the strain of poses generated by docking or co-folding algorithms can be used to distinguish false positives from true binders.
Challenges in Computing Strain
The simplest way to compute strain is simply to run a conformer search and take the difference in energy between the input pose and the global minimum-energy pose. Unfortunately, this approach to strain computation gives bizarre or unphysical energies. Since different levels of theory predict different bond lengths, comparing a conformation produced by e.g. a Vina forcefield to a structure optimized with higher-level methods often predicts vast strain energies (>100 kcal/mol) because each individual bond is predicted to be "strained." While this can be addressed by re-optimizing the entire structure of interest at the chosen level of theory, this quickly becomes expensive and intractable for large systems like protein–ligand complexes, which are exactly the cases where strain calculations are most useful.
Another approach is to simply optimize the input conformation using the chosen level of theory, thus correcting any inaccuracies in bond lengths or angles and leading to lower strain-energy predictions. This can work, but unfortunately often completely changes the overall shape of the conformation—so the resultant strain prediction, while physically valid, isn't even relevant to the input structure. A complete optimization can remove strain from various rotatable dihedrals, even when understanding that strain is the main purpose of the workflow.
Rowan's Approach to Strain Prediction
Rowan's approach to strain prediction uses harmonically restrained optimizations to ensure that the overall molecular conformation is preserved even as individual bonds and angles are allowed to relax. When a strain calculation is requested, Rowan runs an optimization using the same level of theory but augmented with a harmonic restraint that penalizes deviations from the starting coordinates. Heavy atoms (and optionally hydrogens) are kept close to their initial positions via this added quadratic term, so the optimizer can eliminate obvious artifacts—bad angles, clashes, minor distortions—without allowing the structure to collapse into an unrelated conformer. This leads to an approach that's less fragile than running strain without optimization, while avoiding the pathological over-relaxation observed with unconstrained optimization.
Running Strain Predictions in Rowan
Rowan makes it easy to run strain calculations following the above protocol. From Rowan's "strain calculation" workflow, simply upload the desired pose (or draw it using Rowan's 3D editor), choose any level of theory—DFT, semiempirical methods, or neural network potentials—and submit the calculation. Rowan will automatically allocate compute resources, run the calculation, and return results straight to the web browser:
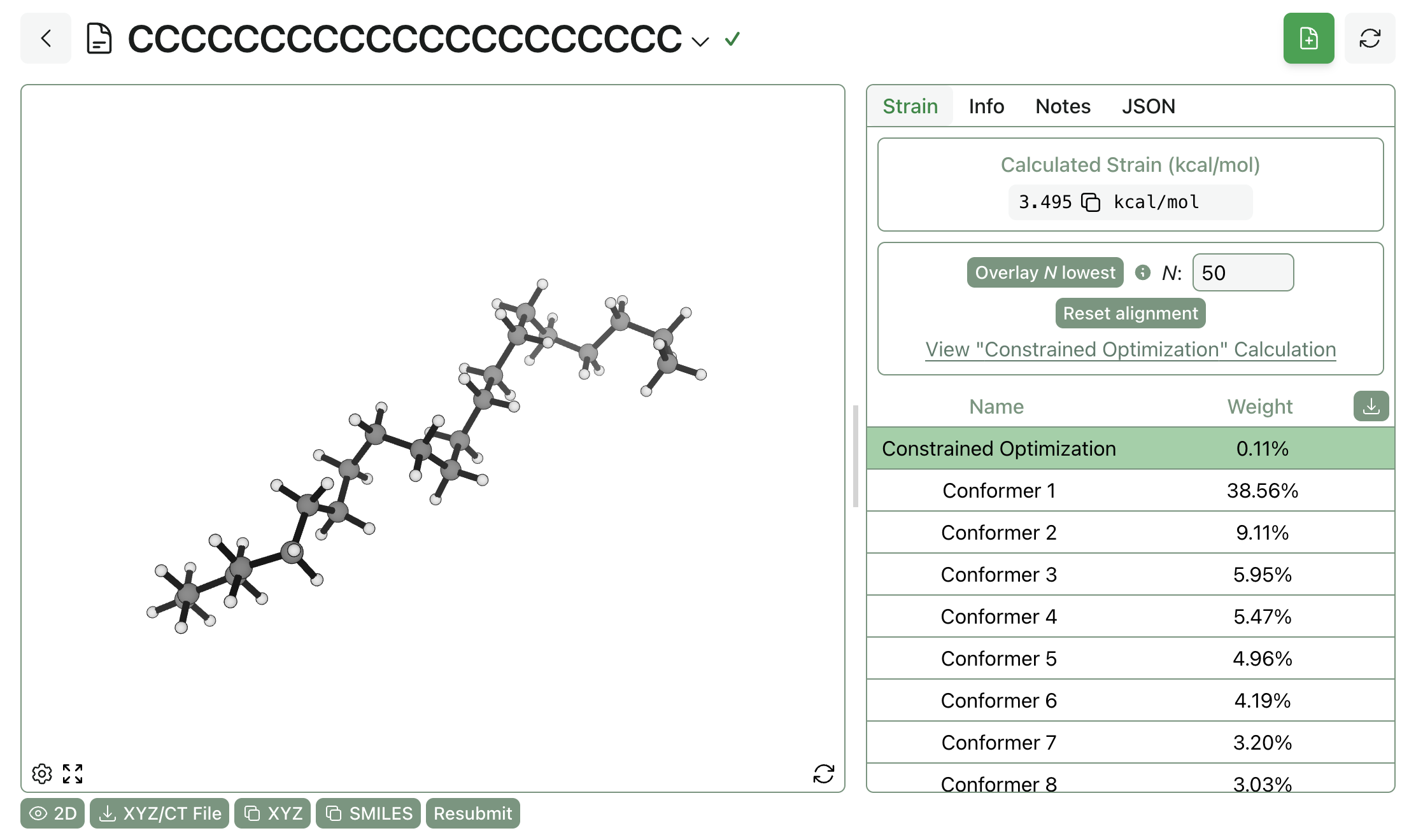
The result of a strain calculation run through Rowan.
It's also easy to run strain calculations using Rowan's free Python API:
from stjames import Molecule
import rowan
# Set ROWAN_API_KEY environment variable to your API key or set rowan.api_key directly
# rowan.api_key = "rowan-sk..."
# ordinarily, we'd try a 3D conformer and see what the strain is.
# here we'll investigate the strain of the autogenerated hexane conformer
result = rowan.submit_strain_workflow(
initial_molecule=Molecule.from_smiles("CCCCCC"),
name="test autogen hexane strain",
)
print(result.wait_for_result().fetch_latest(in_place=True))
