Batch Protein–Ligand Docking
Rowan's batch protein–ligand docking workflow makes it easy to run high-throughput docking calculations or virtual screens against a given ligand target.
Conventional Docking Calculations
Protein-ligand docking calculations can be useful in many contexts. Some docking calculations are run one-at-a-time by medicinal chemists hoping to understand how minute changes to a given lead compound will affect a compound's bound conformation or ligand strain. For cases like this, Rowan's strain-corrected docking workflow is the best choice. This workflow automatically runs careful conformer searches, geometry optimizations with neural network potentials, and PoseBusters- and strain-based checks to ensure maximum physical validity.
Unfortunately, these checks also come at the cost of computational efficiency. Rowan's conventional docking workflow typically takes a few minutes per compound; while this can be decreased to about 20 seconds by tuning settings, this runtime is still too slow for high-throughput virtual screening. It's for situations like this that Rowan's batch docking workflow is designed.
Rowan's Batch Docking Workflow
Rowan's batch docking workflow follows established best practices for optimizing docking runtimes. From a given SMILES string, Rowan quickly adds hydrogen atoms, generates multiple initial conformers, and runs minimal initial optimizations using the MMFF94 force field. The lowest-energy conformer is docked against the specified pocket using QVina2, a high-throughput docking software designed to be significantly faster than AutoDock Vina. To ensure maximum CPU utilization even on large machines, the machine's total core count is detected at runtime and divided into a pool of worker processes; each worker is assigned a fixed number of internal Vina threads, so that multiple ligands are docked in parallel without oversubscribing hardware.
Conventional docking workflows return large amounts of data: each docking run generates multiple poses per compound, and each pose has an associating binding affinity and (optionally) a strain value andan associated refined holo protein–ligand complex. Storing and managing all this data makes sense in a lead-optimization context, but adds significant per-compound overhead. To make running high-throughput virtual screens easier, Rowan's batch docking workflow saves only the best docking score per compound, returning an easy-to-analyze list of data that can be sorted, filtered, and refined with subsequent workflows.
Running Batch Docking in Rowan
Batch docking is available to all organizations using Rowan. To run a batch docking calculation, simply navigate to Rowan's "Batch Docking" workflow, input the protein, pocket, and list of SMILES, and click "Submit." Rowan will automatically allocate a high-performance CPU, run the calculations, and save the results to the database.
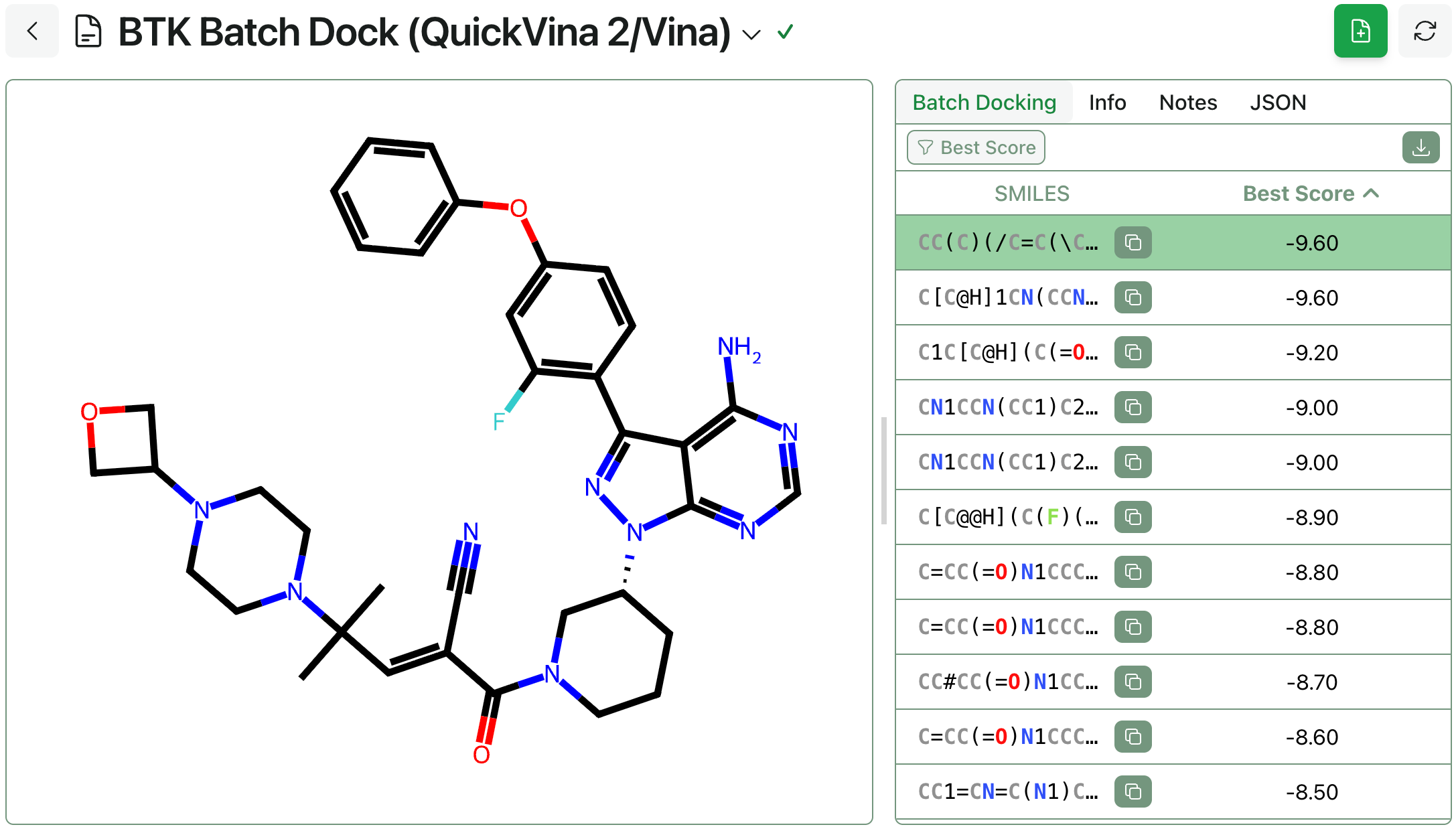
A screenshot of Rowan's batch-docking output.
Batch docking can also be easily run via Rowan's Python API:
import rowan
ligands = ["c1cccnc1CCOCO", ...]
protein_uuid = ...
workflow = rowan.submit_batch_docking_workflow(
ligands,
protein_uuid,
pocket=[[103.55, 100.59, 82.99], [27.76, 32.67, 48.79]],
executable="qvina2",
scoring_function="vina",
)
workflow.wait_for_result().fetch_latest(in_place=True)
print(workflow.data["best_scores"])
