Microscopic pKa Prediction
Knowing a molecule's pKa is fundamental to understanding its structure and properties. The pKa of a molecule determines what its ionization state will be in various media, and thus also dictates a wide variety of important behaviors, like permeability, solubility, and reactivity.
Conventional pKa Calculations
Since experimental pKa measurements are often costly and time-consuming, it's common to use computational tools to get quick estimates of pKa values. Traditional pKa estimates either (1) use empirical data-driven relationships to guess the pKa of unseen compounds by analogy to training data or (2) use high-level quantum chemistry to compute pKa from first principles. These approaches both have advantages and disadvantages: data-driven methods run quickly and display excellent accuracy for compounds similar to those in their training set, but struggle for compounds that they've never seen before, while quantum methods display excellent accuracy but can be very slow (hours or days per calculation).
Rowan's Machine Learning-Based Approach
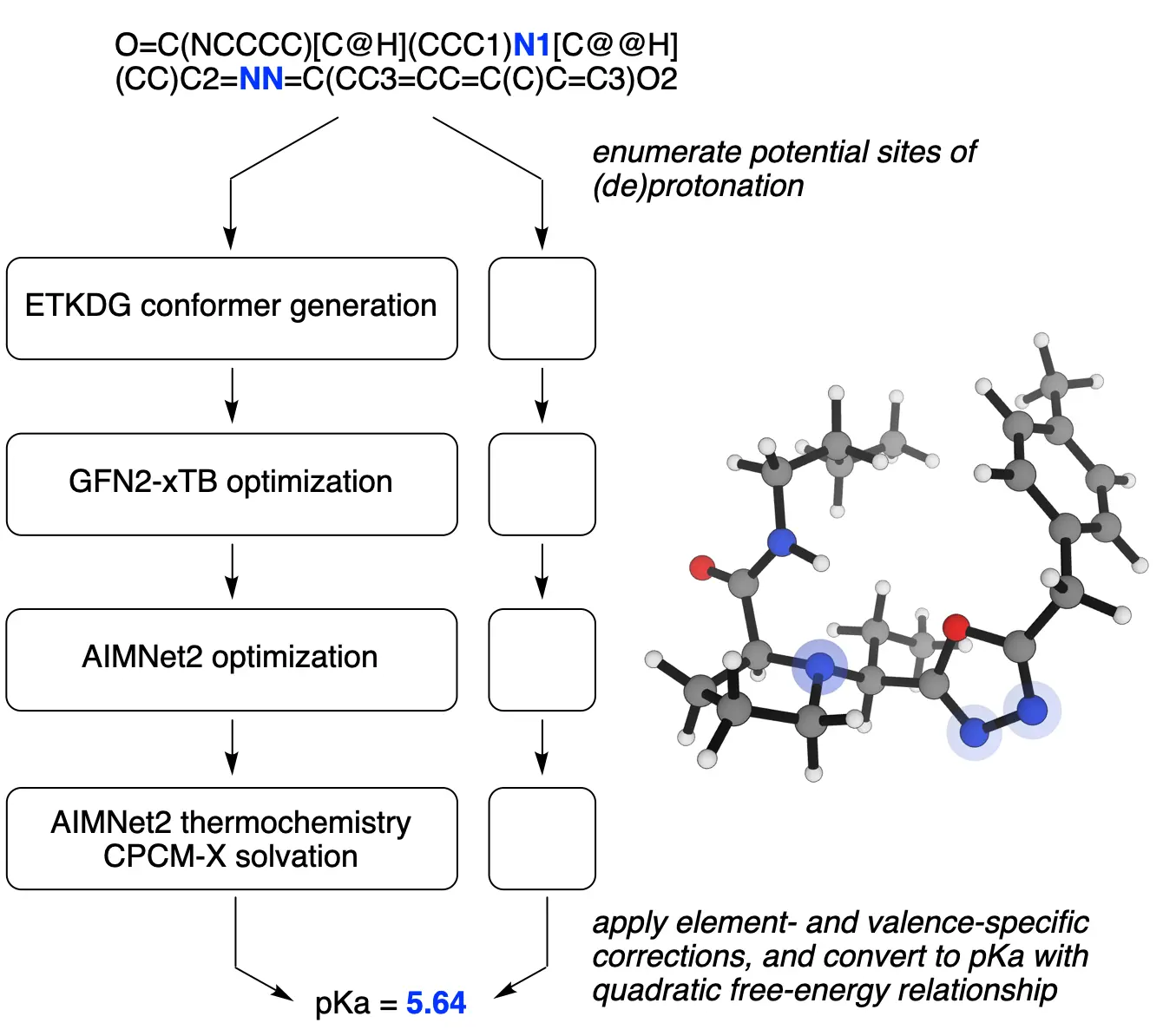
Rowan uses machine learning-based interatomic potentials to vastly accelerate the accurate calculation of pKa values. Our approach uses machine learning as a low-cost way to run physics-based calculations, thus mimicking the accuracy of quantum mechanics-based modeling for a tiny fraction of the cost. On a database of 100 medicinally relevant ring systems, Rowan's pKa workflow took an average of only 23 seconds per molecule to compute all the sites of protonation and deprotonation! We've validated the accuracy of Rowan's pKa prediction workflow on a variety of challenging test sets: see our preprint for a full assessment.
Key Advantages of Rowan pKa
Rowan's pKa prediction workflow runs quickly, can operate on a variety of file formats (including text representations like SMILES strings), and generates an intuitive, publication-quality output that clearly highlights potential sites of protonation and deprotonation on the input molecule. Here's what the output of Rowan's pKa workflow looks like:
Rowan also computes and displays the geometry of the conjugate acids and bases generated by (de)protonation, allowing for visualization of conformational changes and analysis of the factors leading to attenuated/enhanced acidity and basicity.
