Pose-Analysis Molecular Dynamics
Most structure-based-drug-design programs start with methods that can generate protein–ligand complexes, like docking or newer co-folding algorithms. These methods are the mainstay of much virtual screening work, and Rowan's put a considerable amount of effort into making sure our docking and co-folding workflows run quickly and securely while producing physically valid results.
It's often the case, however, that scientists want to study more than just a static predicted protein–ligand complex. In these cases, it's helpful to run molecular dynamics (MD) simulations on the predicted binding pose: viewing and analyzing the resultant trajectory can yield important insights into individual inner-sphere interactions or amino-acid movement. Work from Hugo Guterres and Wonpil Im also suggests that simply analyzing the pose stability from MD could serve as an effective post-docking filter in some cases.
Pose-Analysis Molecular Dynamics in Rowan
Rowan's pose-analysis MD workflow can be used to run simulations of protein–ligand complexes. Starting from a refined holo protein–ligand complex (like that generated by Rowan's docking or co-folding workflows), a custom small-molecule forcefield is automatically built using OpenForceField (Sage).
System preparation is done automatically. Explicit waters and ions are added to solvate the system to, by default, an ionic strength of 0.10 M. Protein backbone constraints are added for residues more than 7.0 Å away to prevent any unfolding; hydrogen mass is increased to 1.5 Daltons and bonds to hydrogen are constrained, allowing OpenMM to take 2.0 fs timesteps; and the particle-mesh Ewald algorithm is used to handle long-range interactions with a 8.0 Å cutoff distance. (All these parameters are configurable via Rowan's Python API.)
Calculations are run in the NPT ensemble using a Langevin thermostat (1 ps timescale) and a Monte Carlo barostat. The temperature and pressure are 300 K and 1 atm by default. After minimization and gradually heating the system to the desired temperature, a 1 ns equilibration simulation is run followed by a user-specified length and number of replicas of the "production" simulation.
Running Molecular Dynamics in Rowan
Rowan makes it easy to run pose-analysis MD simulations. From the web, simply upload or resubmit a protein–ligand complex, select the "Pose Analysis MD" workflow, and choose the relevant run parameters.
The workflow will run on an automatically allocated GPU and, when complete, display the trajectory on the web. Here's a graph of ligand RMSD by frame through Rowan's web viewer:
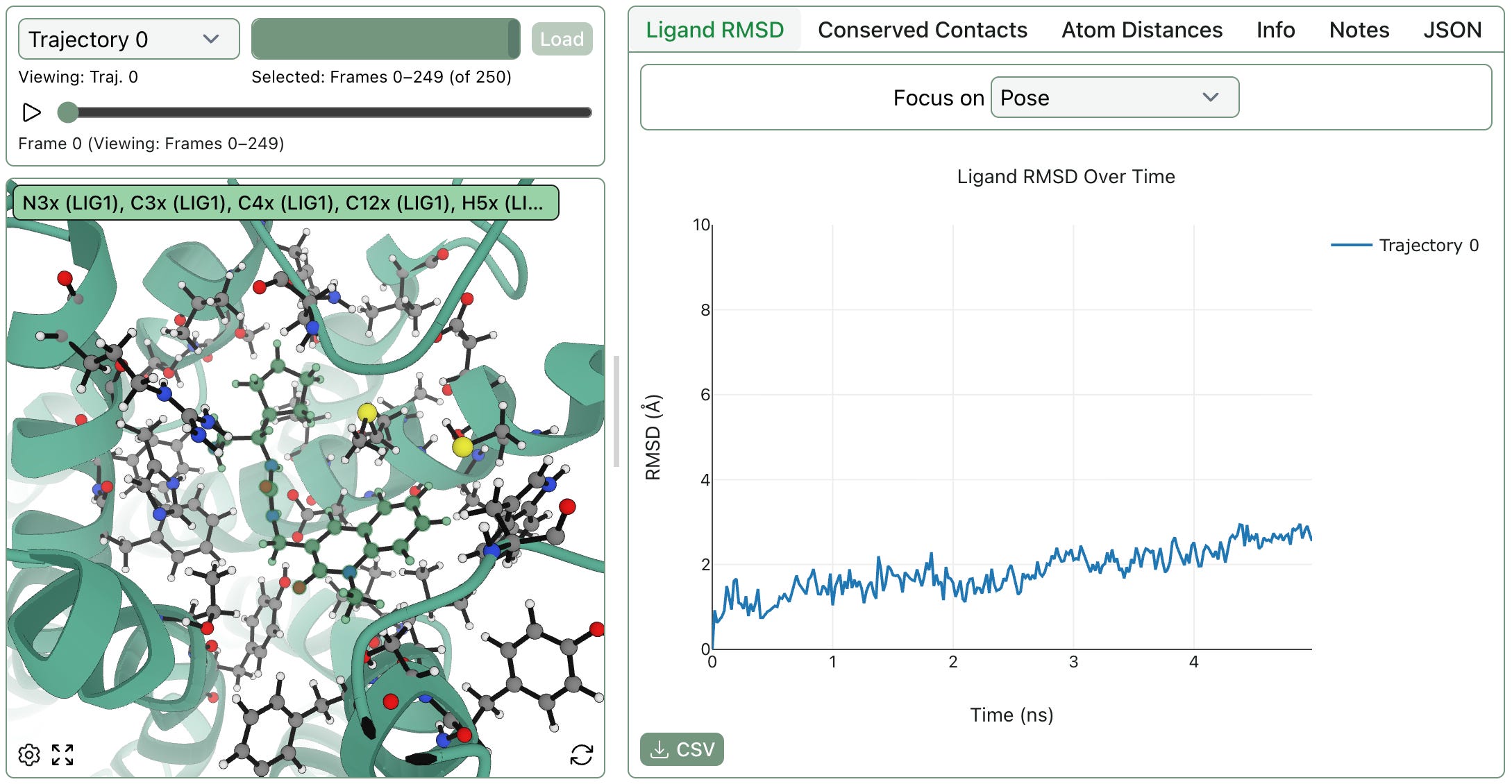
A screenshot of Rowan's pose-analysis MD results.
Pose-analysis MD can also be run via Rowan's Python API. The below script automatically runs a pose-analysis MD simulation on the result of a co-folding workflow:
import rowan
ligand = "CCC(C)(C)NC1=NCC2(CCC(=O)C2C)N1"
cofolding_workflow = rowan.submit_protein_cofolding_workflow(
initial_protein_sequences=[
"MENFQKVEKIGEGTYGVVYKARNKLTGEVVALKKIRLDTETEGVPSTAIREISLLKELNHPNIVKLLDVIHTENKLYLVFEFLHQDLKKFMDASALTGIPLPLIKSYLFQLLQGLAFCHSHRVLHRDLKPQNLLINTEGAIKLADFGLARAFGVPVRTYTHEVVTLWYRAPEILLGCKYYSTAVDIWSLGCIFAEMVTRRALFPGDSEIDQLFRIFRTLGTPDEVVWPGVTSMPDYKPSFPKWARQDFSKVVPPLDEDGRSLLSQMLHYDPNKRISAKAALAHPFFQDVTKPVPHLRL"
],
initial_smiles_list=[ligand],
ligand_binding_affinity_index=0,
name=f"Cofolding {ligand}",
do_pose_refinement=True,
)
cofolding_workflow.wait_for_result().fetch_latest(in_place=True)
md_workflow = rowan.submit_pose_analysis_md_workflow(
protein=cofolding_workflow.data["predicted_refined_structure_uuid"],
initial_smiles=ligand,
num_trajectories=1,
simulation_time_ns=1,
name="Downstream molecular dynamics",
)
md_workflow.wait_for_result().fetch_latest(in_place=True)
# print ligand RMSD by frame
print(md_workflow.data["trajectories"][0]["rmsd"])
