Modeling Complexes and Ranking Ligands with Boltz-2
For a comparison against Rowan's other co-folding models, see our co-folding overview page.
Most protein–ligand structure-prediction tools, like AlphaFold 3, Boltz-1, and Chai-1, stop at predicting the geometry of the resultant complex. Boltz-2 (preprint) goes further by predicting both structures and small-molecule binding affinity from sequence and SMILES, while supporting DNA and RNA partners. It builds on the cofolding ideas in Boltz-1, then adds a larger backbone, better controllability, and a dual affinity head. The result is a practical way to generate poses, prioritize compounds, and guide follow-up physics or experiments when speed matters.
What's New in Boltz-2
Boltz-2 is similar to Boltz-1, but contains numerous enhancements and new capabilities. Here's a brief overview:
| Aspect | Boltz-1 | Boltz-2 |
|---|---|---|
| Structural engine | 48 PairFormer layers, 512-token crop | 64 layers, trifast kernels, 768-token crop |
| Controllability | None | • Method conditioning (X-ray / NMR / MD) • Multi-chain template steering • Contact & pocket constraints |
| Physics quality | Optional steering potential (Boltz-1x) | Optional steering potential (Boltz-2x) |
| Affinity head | None | PairFormer-based dual head (probability + pIC50) |
| MSA | Optional | De facto required |
Here's the authors' architecture diagram:
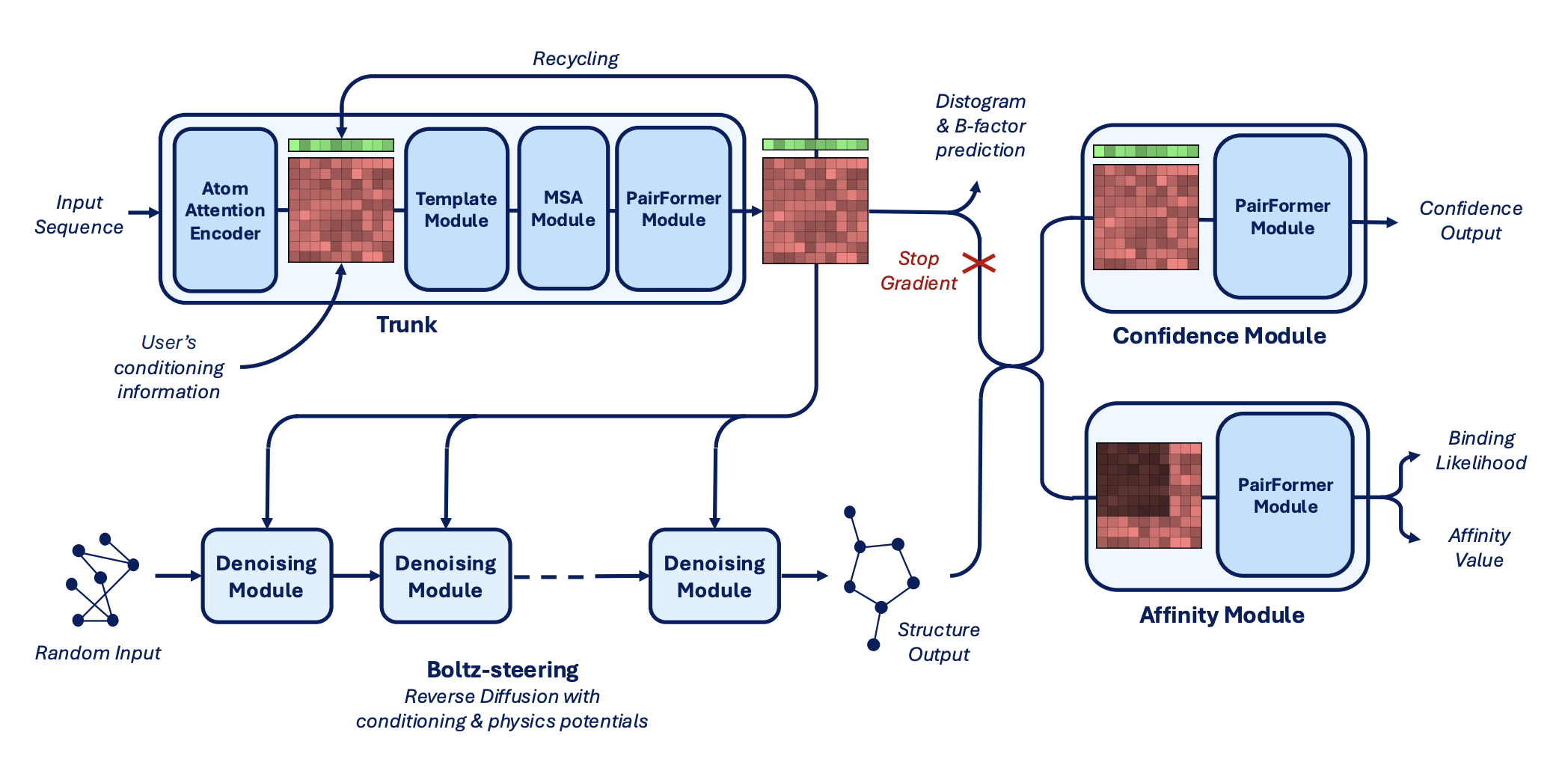
The architecture of Boltz-2 (Figure 2 from the preprint).
Several of these new features are incredibly powerful:
- Affinity outputs. Two signals are produced for protein–ligand pairs: a classifier for binder vs decoy that works well for triaging screens, and a quantitative affinity head that estimates pIC50 for SAR ranking.
- Method conditioning. Users can nudge sampling toward structural states typical of X-ray, cryo-EM, solution NMR, MD and more. This helps align results with your intended experimental context.
- Template and constraints. Lock specific chains near a structural template—uploaded directly or pulled in by PDB ID—or use contact and pocket constraints to keep key residues in play. Template influence is tunable, from a soft structural prior up to a nearly fixed scaffold; see our co-folding overview for how this works and a runnable example. (OpenFold-3 supports templates too, if you'd like to compare.)
- Optional steering potentials. Inference-time steering potentials (Boltz-2x) that can reduce clashes without heavy relaxation, albeit at the cost of slower inference.
For more details about how Boltz-2 works, see our full Boltz-2 FAQ or the original preprint.
As of June 2026, the latest Boltz-2 model, "Boltz 2.1," is closed-source and can only be run via the Boltz-hosted API. Rowan users can run Boltz 2.1 using the same interface as previous Boltz models, but all co-folding jobs will be run on Boltz servers and subject to their terms of service.
Where Boltz-2 Can Be Useful
Cofolding is useful whenever a rigid receptor limits realism. Boltz-2 generates binding poses that include side chain and backbone motion, then attaches a confidence score and an affinity estimate. This is not a replacement for free-energy perturbation or long-timescale MD, but it is a strong first pass that can prune search space and surface plausible chemotypes.
- Hit discovery. Use the binder probability to sort very unbalanced libraries before docking or physics.
- Lead optimization. Use the pIC50 head to rank close analogs and decide what to simulate or synthesize.
- Difficult pockets. Cryptic or flexible sites benefit from joint sampling of protein and ligand.
Here's a plot showing Boltz-2's accuracy in virtual screening, illustrating superior accuracy vs. other high-throughput binding-affinity-prediction methods:
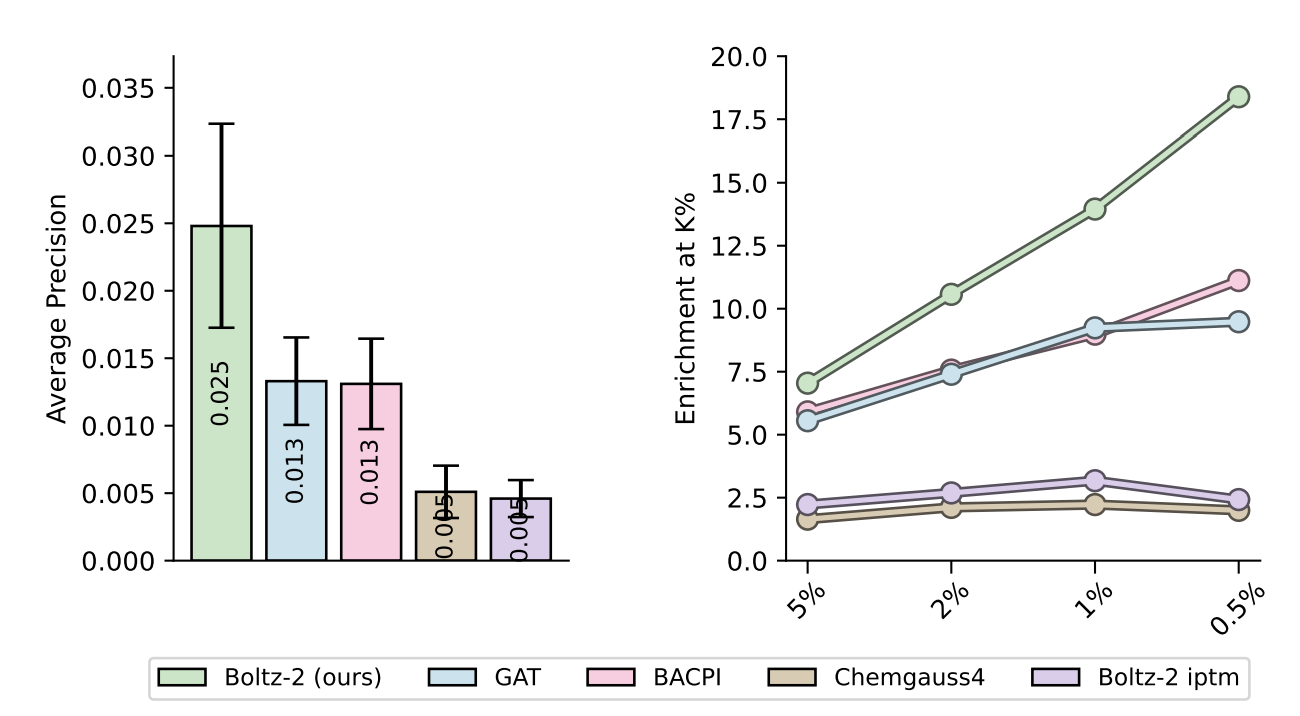
Figure 7 from the Boltz-2 paper, comparing Boltz-2's affinity prediction to other low-cost methods.
For more on the strengths of Boltz-2 and limitations vs. other binding-affinity-prediction methods, see our page comparing different methods for predicting binding affinity.
Limitations of Boltz-2
Despite the myriad successes of Boltz-2, challenges remain:
- Small-molecule conformations and stereochemistry are still often predicted incorrectly: see inter alia this Github issue.
- Allosteric binders are not described well; see this benchmarking work from Pat Walters.
- Affinity from public data is noisy, and large conformational changes can still fool any single pass predictor. Furthermore, membrane targets like GPCRs and channels show higher variance.
- Metal coordination, ordered waters, or multimeric cofactors are not modeled well by the current affinity head.
- And Boltz-2 doesn't output hydrogen positions, making it somewhat difficult to run molecular dynamics or other rescoring methods directly on output files.
Furthermore, there are concerns that the reported performance of Boltz-2 can in large part be attributed to train–test leakage, meaning that the performance on targets dissimilar to the training set will be much worse. (Here's a review from Karson Crispens and co-workers which alleges this.)
For a compilation of Boltz-2 benchmark results, see our external Boltz-2 benchmark page.
Using Boltz-2 on Rowan
Rowan makes it straightforward to integrate Boltz-2 and other co-folding methods into modern drug-discovery teams. Through Rowan's easy-to-use web interface, scientists can upload sequences and ligands, submit Boltz-2 jobs to GPU runners and our privately hosted secure MSA server, and analyze the resulting structures and predicted binding affinity. Rowan automatically applies a series of structural checks and filters to help scientists detect model failures or invalid results and focus only on the most trustworthy predictions.
Here's what a completed Boltz-2 prediction looks like on Rowan. The below ligand did not pass the automatic PoseBuster screen, indicating that the predicted pose is unphysical and might not be trustworthy. The ligand's strain is predicted to be 4.17 kcal/mol, which is a little high but not too high to be reasonable. For chiral ligands, an automatic stereochemistry check is also applied, which helps to screen out cases in which diffusion steps incorrectly invert stereocenters.
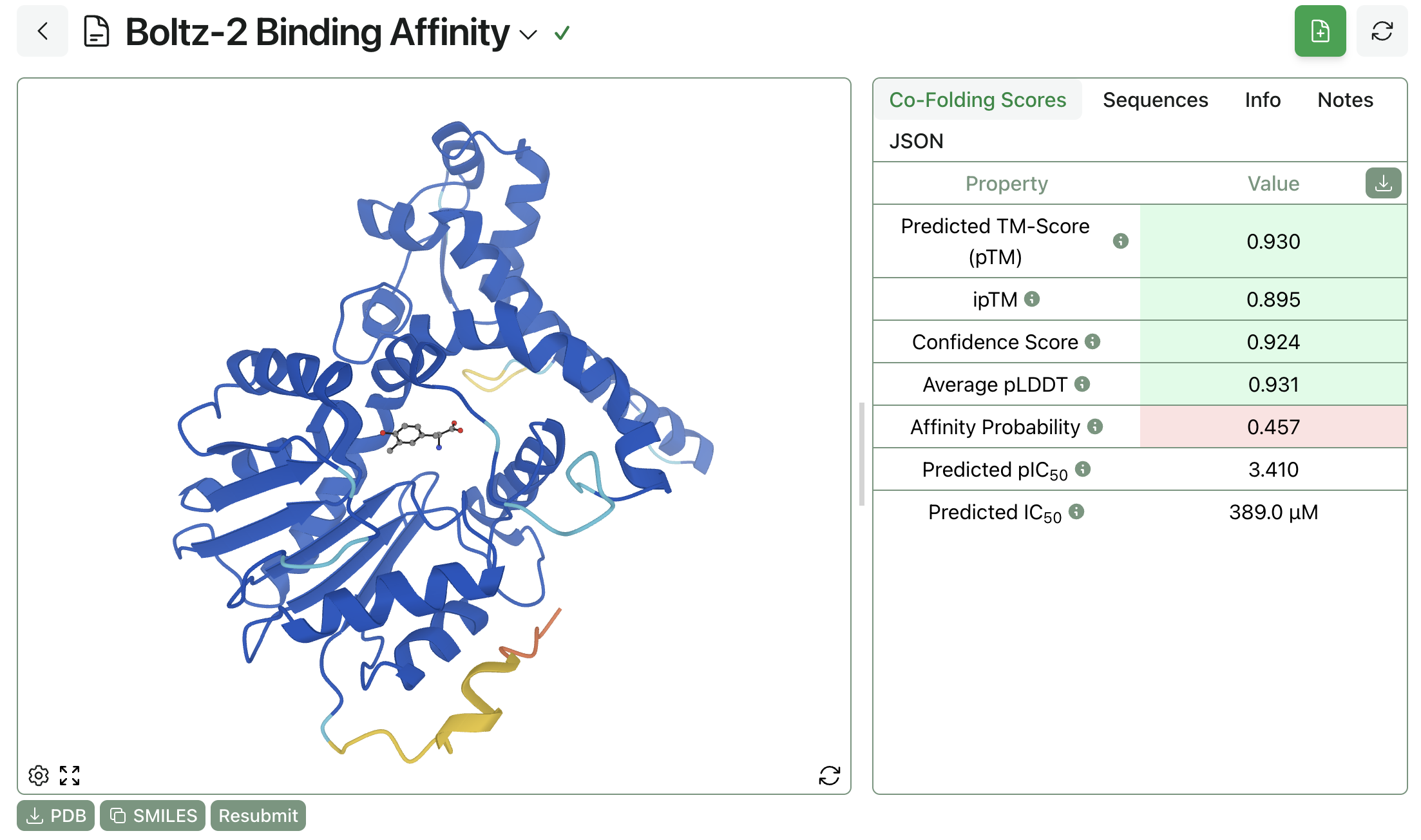
Rowan also makes it easy to validate Boltz-2 results with other methods, including Chai-1, docking, and molecular dynamics. From a completed Boltz-2 workflow, it's easy to resubmit structures to any other Rowan-supported method and compare the completed results to Boltz-2 structures. Although native Boltz-2 predictions lack most ligand hydrogens, making it difficult to submit Boltz-2 structures to other workflows, Rowan automatically adds hydrogen atoms to make downstream simulation seamless.
Python API
Boltz-2 calculations can also easily be submitted via Rowan's Python API, which is available for free to all Rowan users:
import rowan
rowan.api_key = "rowan-sk-asdf"
workflow = rowan.submit_protein_cofolding_workflow(
initial_protein_sequences=[
"MENFQKVEKIGEGTYGVVYKARNKLTGEVVALKKIRLDTETEGVPSTAIREISLLKELNHPNIVKLLDVIHTENKLYLVFEFLHQDLKKFMDASALTGIPLPLIKSYLFQLLQGLAFCHSHRVLHRDLKPQNLLINTEGAIKLADFGLARAFGVPVRTYTHEVVTLWYRAPEILLGCKYYSTAVDIWSLGCIFAEMVTRRALFPGDSEIDQLFRIFRTLGTPDEVVWPGVTSMPDYKPSFPKWARQDFSKVVPPLDEDGRSLLSQMLHYDPNKRISAKAALAHPFFQDVTKPVPHLRL"
],
initial_smiles_list=["CCC(C)CN=C1NCC2(CCCOC2)CN1"],
name="Boltz-2 cofolding job",
model="boltz_2",
ligand_binding_affinity_index=0,
)
workflow.wait_for_result()
workflow.fetch_latest(in_place=True)
print(workflow.data)
print(f"View the 3D structure on the web at labs.rowansci.com/protein-cofolding/{workflow.uuid}")
This prints the following data; the corresponding 3D structure can be viewed through Rowan's web interface.
{
'lddt': [0.779, 0.909, 0.958, 0.982, 0.979, 0.969, 0.975, 0.959, 0.952, 0.918, 0.885, 0.848, 0.828, 0.803, 0.824, 0.873, 0.912, 0.953, 0.975, 0.985, 0.987, 0.987, 0.978, 0.969, 0.928, 0.946, 0.976, 0.961, 0.984, 0.987, 0.979, 0.977, 0.959, 0.918, 0.889, 0.738, 0.69, 0.614, 0.533, 0.575, 0.578, 0.707, 0.763, 0.809, 0.841, 0.877, 0.904, 0.896, 0.923, 0.948, 0.941, 0.947, 0.952, 0.953, 0.963, 0.962, 0.961, 0.929, 0.969, 0.988, 0.988, 0.99, 0.986, 0.985, 0.985, 0.981, 0.98, 0.971, 0.967, 0.962, 0.909, 0.865, 0.825, 0.766, 0.864, 0.915, 0.964, 0.972, 0.983, 0.979, 0.965, 0.986, 0.985, 0.952, 0.976, 0.972, 0.989, 0.973, 0.973, 0.989, 0.987, 0.982, 0.981, 0.984, 0.977, 0.976, 0.964, 0.986, 0.989, 0.987, 0.989, 0.989, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.989, 0.989, 0.99, 0.988, 0.983, 0.989, 0.987, 0.976, 0.965, 0.974, 0.953, 0.979, 0.951, 0.95, 0.985, 0.972, 0.985, 0.945, 0.914, 0.989, 0.985, 0.989, 0.988, 0.986, 0.977, 0.989, 0.989, 0.99, 0.989, 0.987, 0.971, 0.906, 0.883, 0.876, 0.894, 0.882, 0.877, 0.845, 0.838, 0.814, 0.82, 0.779, 0.752, 0.776, 0.746, 0.678, 0.744, 0.783, 0.766, 0.808, 0.81, 0.928, 0.964, 0.983, 0.987, 0.975, 0.989, 0.989, 0.989, 0.978, 0.987, 0.99, 0.984, 0.979, 0.95, 0.967, 0.939, 0.986, 0.988, 0.99, 0.987, 0.989, 0.99, 0.99, 0.989, 0.99, 0.99, 0.99, 0.99, 0.99, 0.99, 0.989, 0.99, 0.989, 0.988, 0.987, 0.985, 0.986, 0.989, 0.989, 0.985, 0.983, 0.948, 0.967, 0.969, 0.987, 0.985, 0.986, 0.988, 0.989, 0.989, 0.99, 0.99, 0.989, 0.989, 0.989, 0.989, 0.99, 0.989, 0.988, 0.989, 0.986, 0.986, 0.99, 0.989, 0.989, 0.989, 0.989, 0.985, 0.987, 0.987, 0.988, 0.989, 0.989, 0.988, 0.989, 0.989, 0.989, 0.988, 0.989, 0.986, 0.986, 0.981, 0.95, 0.93, 0.974, 0.982, 0.979, 0.987, 0.974, 0.897, 0.989, 0.984, 0.985, 0.989, 0.99, 0.988, 0.988, 0.99, 0.99, 0.987, 0.99, 0.99, 0.99, 0.989, 0.989, 0.99, 0.989, 0.988, 0.987, 0.99, 0.99, 0.99, 0.99, 0.989, 0.99, 0.99, 0.99, 0.989, 0.99, 0.989, 0.99, 0.99, 0.987, 0.988, 0.989, 0.989, 0.988, 0.987, 0.972, 0.984, 0.945, 0.966, 0.901, 0.929, 0.502, 0.601, 0.671, 0.589, 0.677, 0.666, 0.735, 0.695, 0.671, 0.801, 0.71, 0.707, 0.733, 0.743, 0.738, 0.709, 0.69],
'model': 'boltz_2',
'scores': {'ptm': 0.974, 'iptm': 0.87, 'avg_lddt': 0.936, 'confidence_score': 0.923},
'messages': [],
'affinity_score': {'pred_value': 1.434, 'pred_value1': 2.226, 'pred_value2': 0.643, 'probability_binary': 0.045, 'probability_binary1': 0.034, 'probability_binary2': 0.056},
'use_msa_server': True,
'use_potentials': False,
'pocket_constraints': [],
'contact_constraints': [],
'initial_smiles_list': ['CCC(C)CN=C1NCC2(CCCOC2)CN1'],
'use_templates_server': False,
'predicted_structure_uuid': '7eb21671-eec8-450a-a64c-fbb46fdd758d',
'initial_protein_sequences': ['MENFQKVEKIGEGTYGVVYKARNKLTGEVVALKKIRLDTETEGVPSTAIREISLLKELNHPNIVKLLDVIHTENKLYLVFEFLHQDLKKFMDASALTGIPLPLIKSYLFQLLQGLAFCHSHRVLHRDLKPQNLLINTEGAIKLADFGLARAFGVPVRTYTHEVVTLWYRAPEILLGCKYYSTAVDIWSLGCIFAEMVTRRALFPGDSEIDQLFRIFRTLGTPDEVVWPGVTSMPDYKPSFPKWARQDFSKVVPPLDEDGRSLLSQMLHYDPNKRISAKAALAHPFFQDVTKPVPHLRL'],
'ligand_binding_affinity_index': 0
}
For a more complex example showing how to do a virtual screen in parallel with Boltz-2, see our example script on GitHub. Rowan automatically scales GPU-based compute to meet demand, making it possible to run hundreds or thousands of Boltz-2 jobs in just a few lines of code.
Ready to test a target or a series? Create a free Rowan account and start running Boltz-2 today.
