Modern tools for structure-based drug design
Go from protein structure to prioritized compounds with docking, simulation, and binding-affinity workflows in one modern platform.
Go from protein structure to prioritized compounds with docking, simulation, and binding-affinity workflows in one modern platform.

For biotech teams that want trustworthy computational support without assembling and maintaining a stack of disconnected tools.
From first structure to lead optimization, Rowan helps teams:
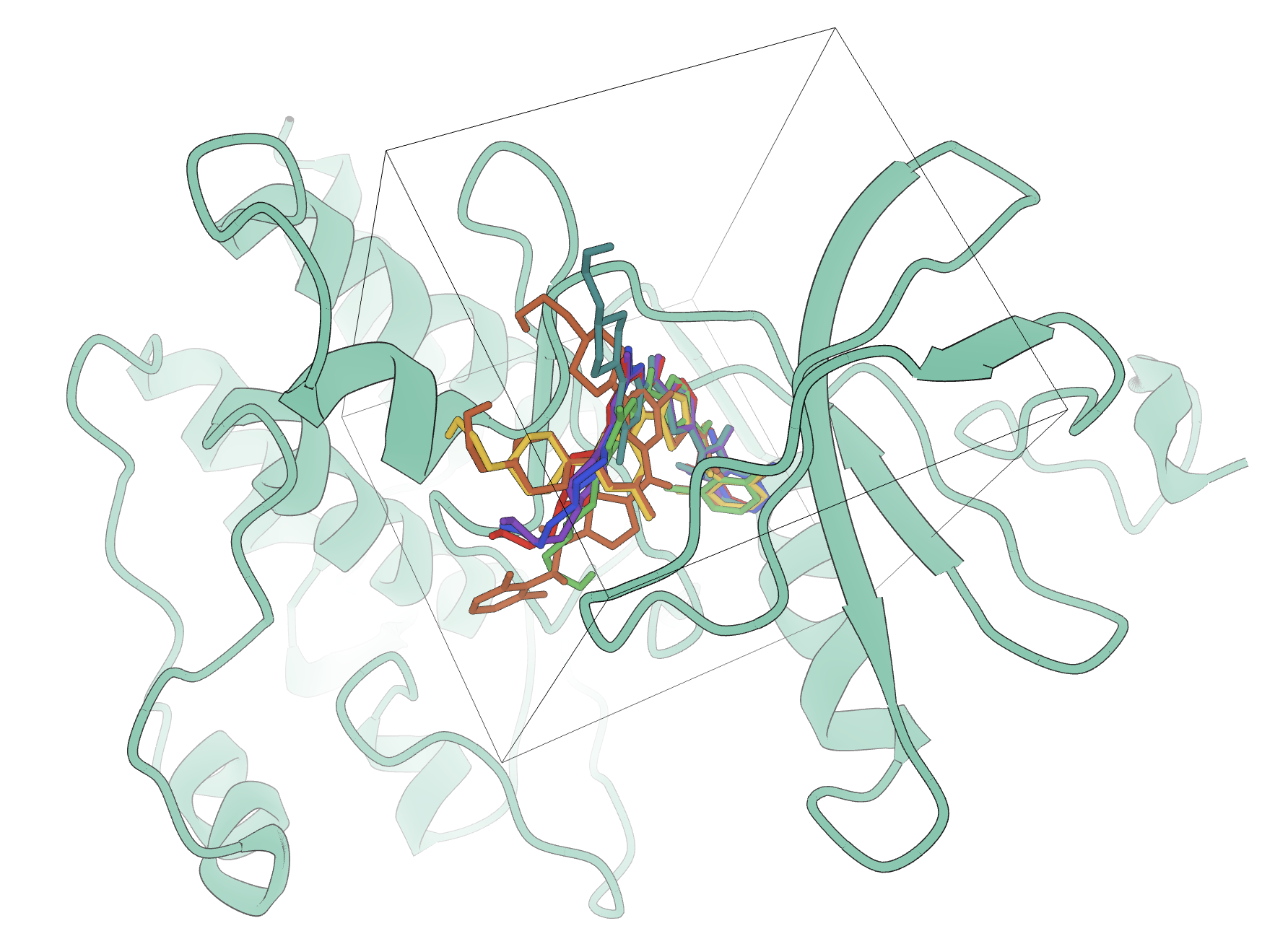
Interrogate protein–ligand binding with docking workflows, short-timescale MD, and interaction analysis tools. Study binding modes, specific contacts, and pose stability in one place.
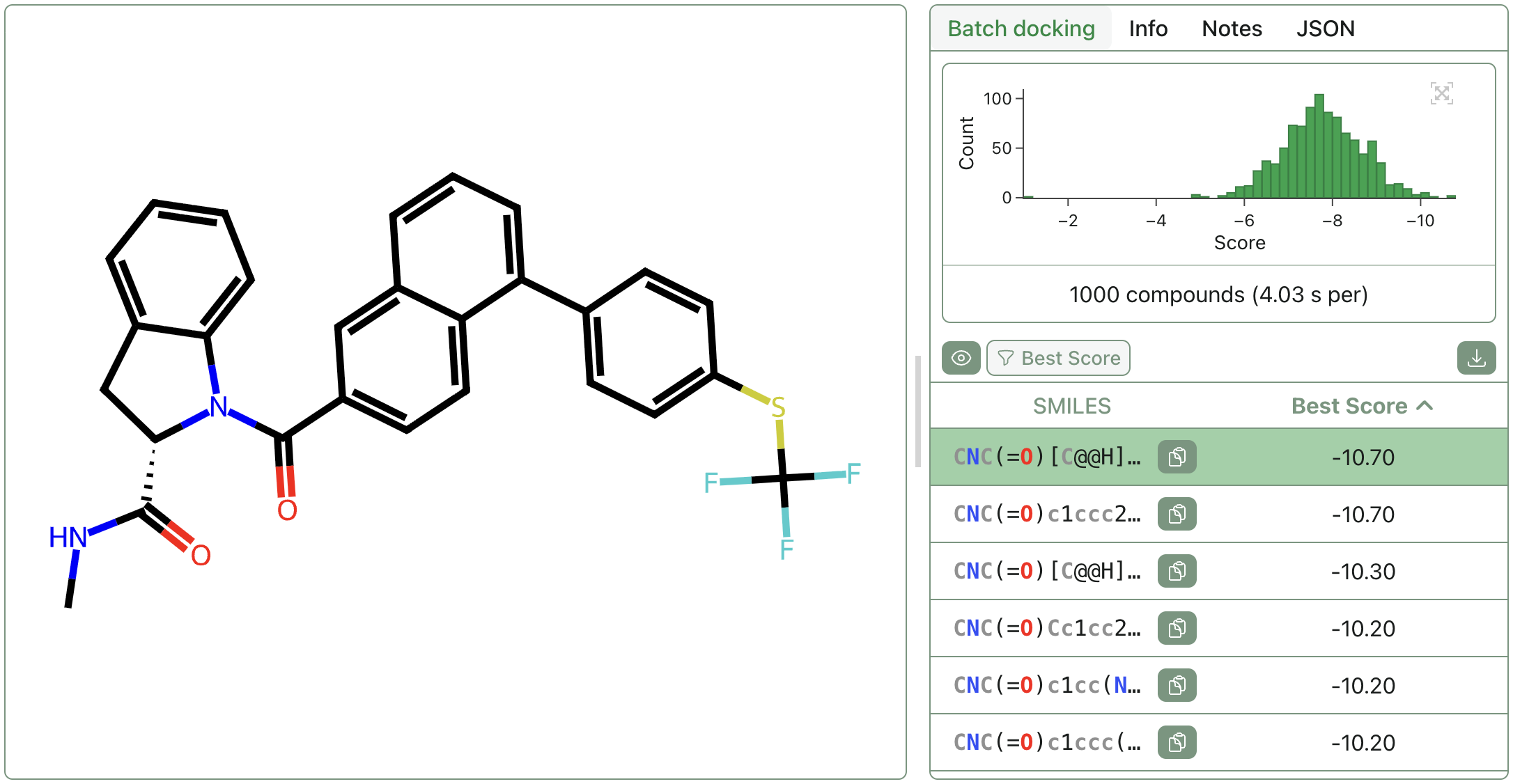
Screen libraries of compounds against a pocket with Rowan's high-speed batch-docking workflow.
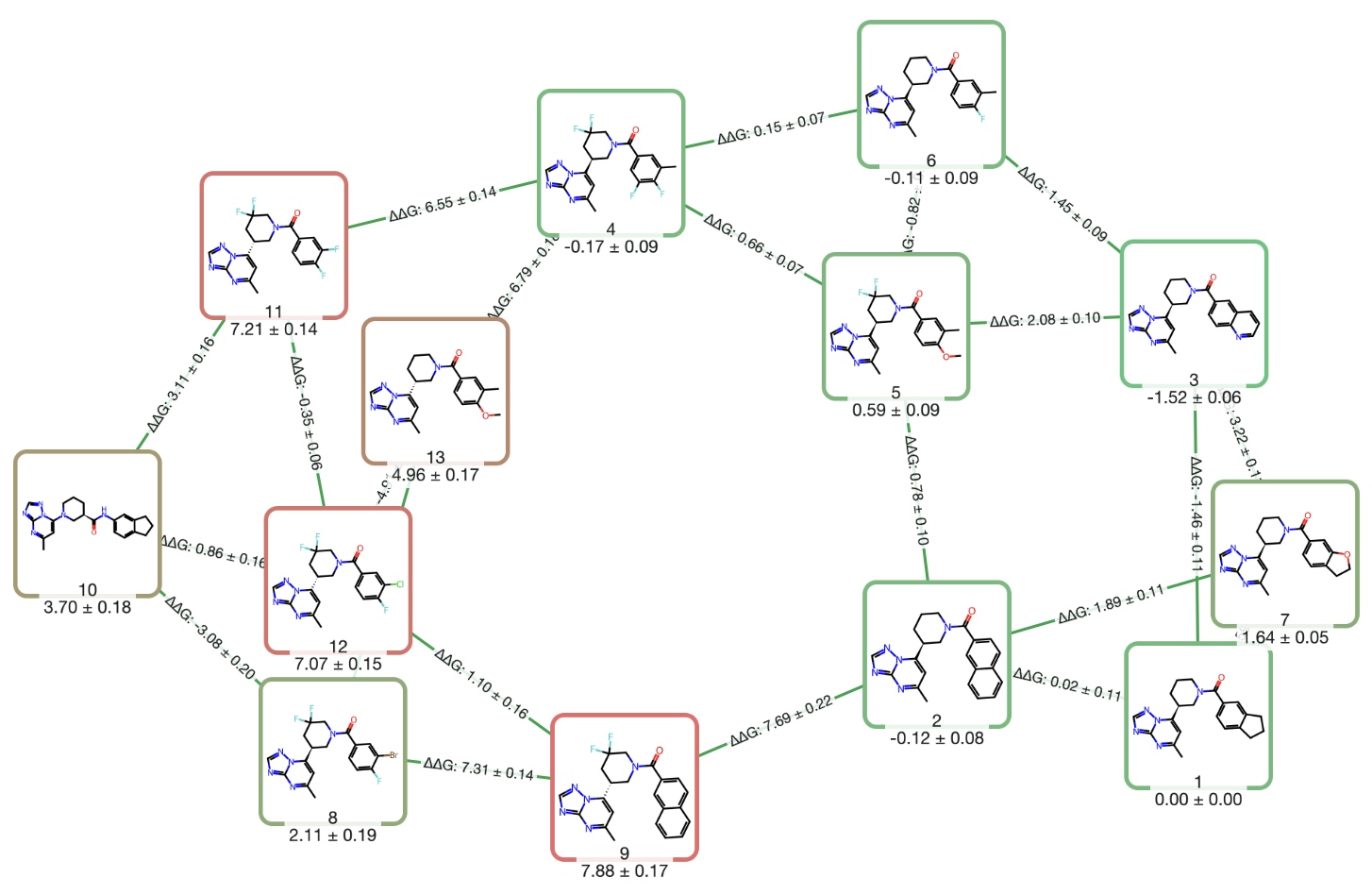
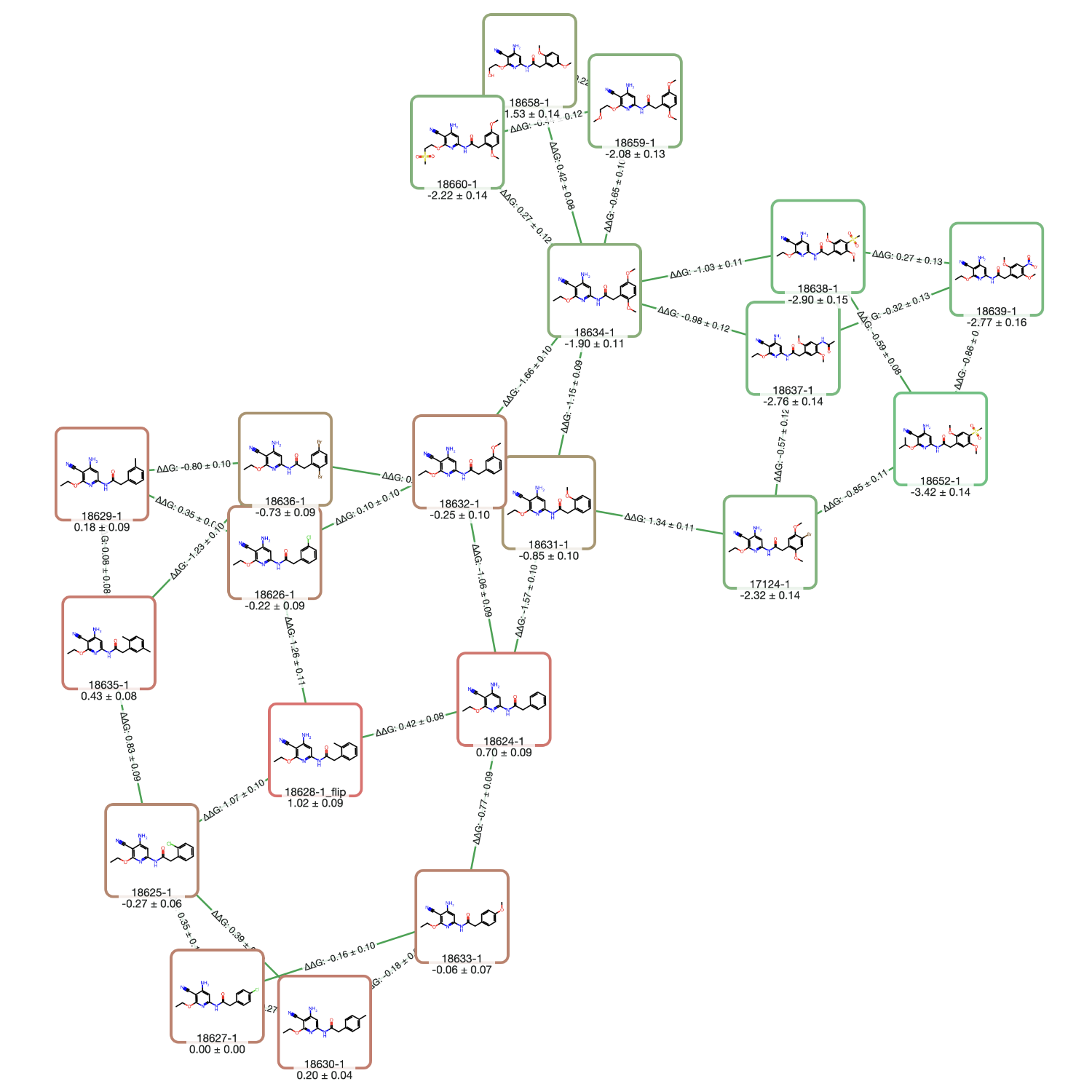
Use RBFE calculations to prioritize analogs, estimate affinity shifts, and support potency & selectivity optimization.
Learn more →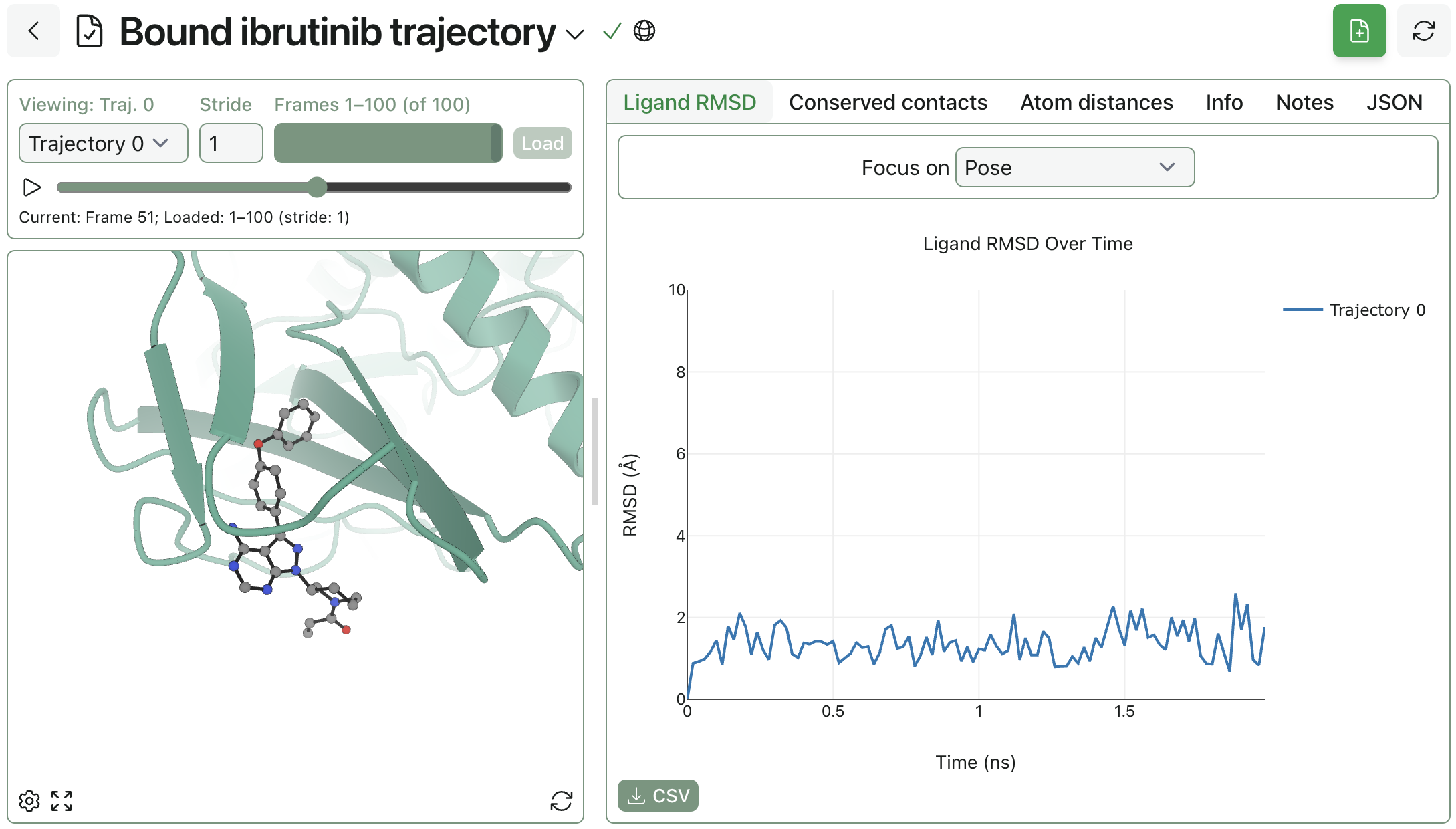
Inspect protein structures, binding poses, conformer ensembles, and MD trajectories directly in the browser, then securely share results with your team.
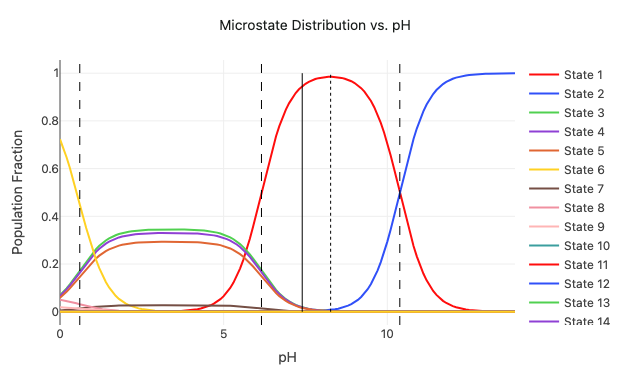
Predict ligand properties such as pKa, solubility, permeability, and solvent-dependent conformer populations to support structure-based design decisions.
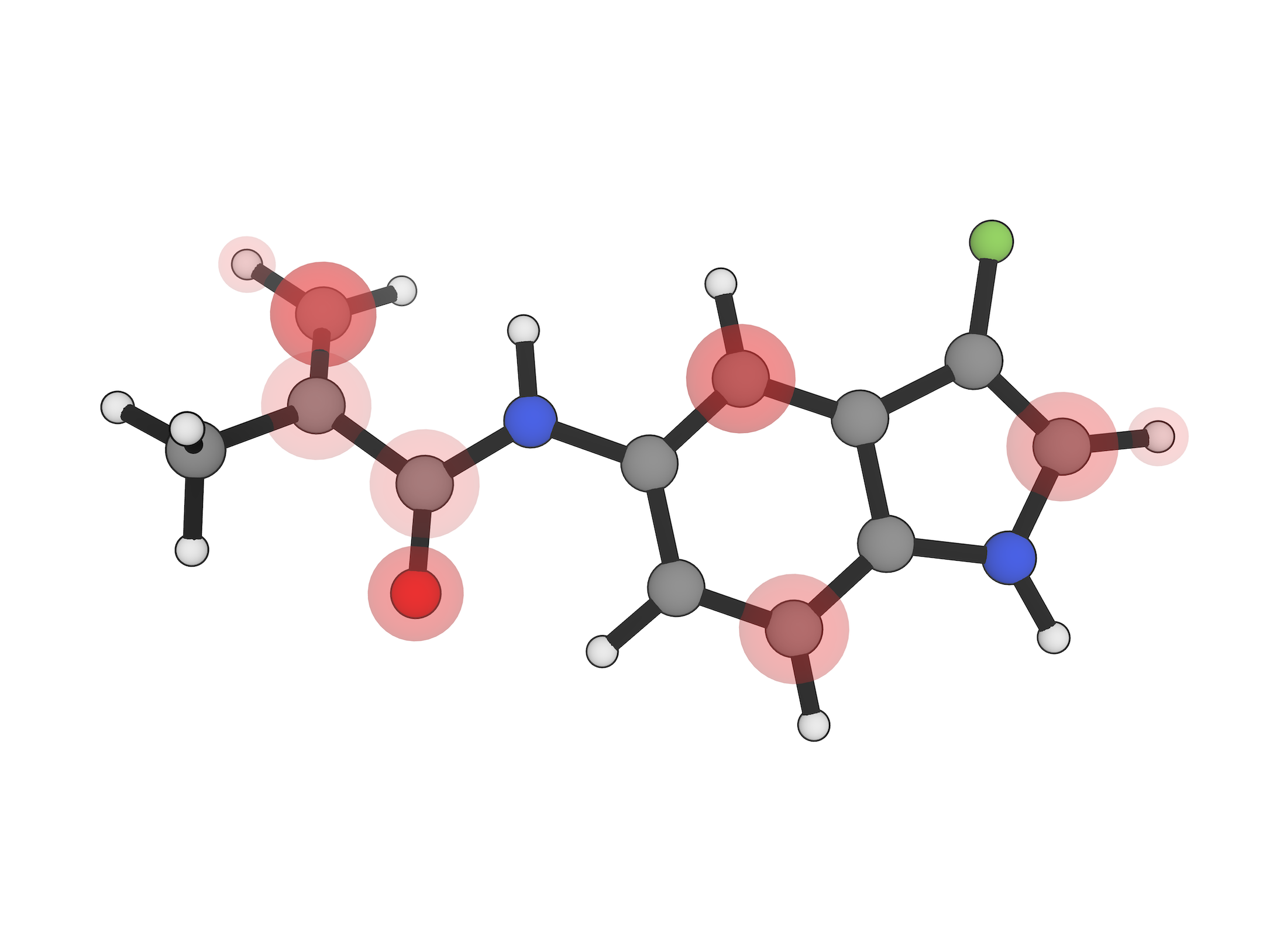
Evaluate covalent warhead reactivity with Fukui indices, transition-state modeling, and reaction-path analysis.
Learn more →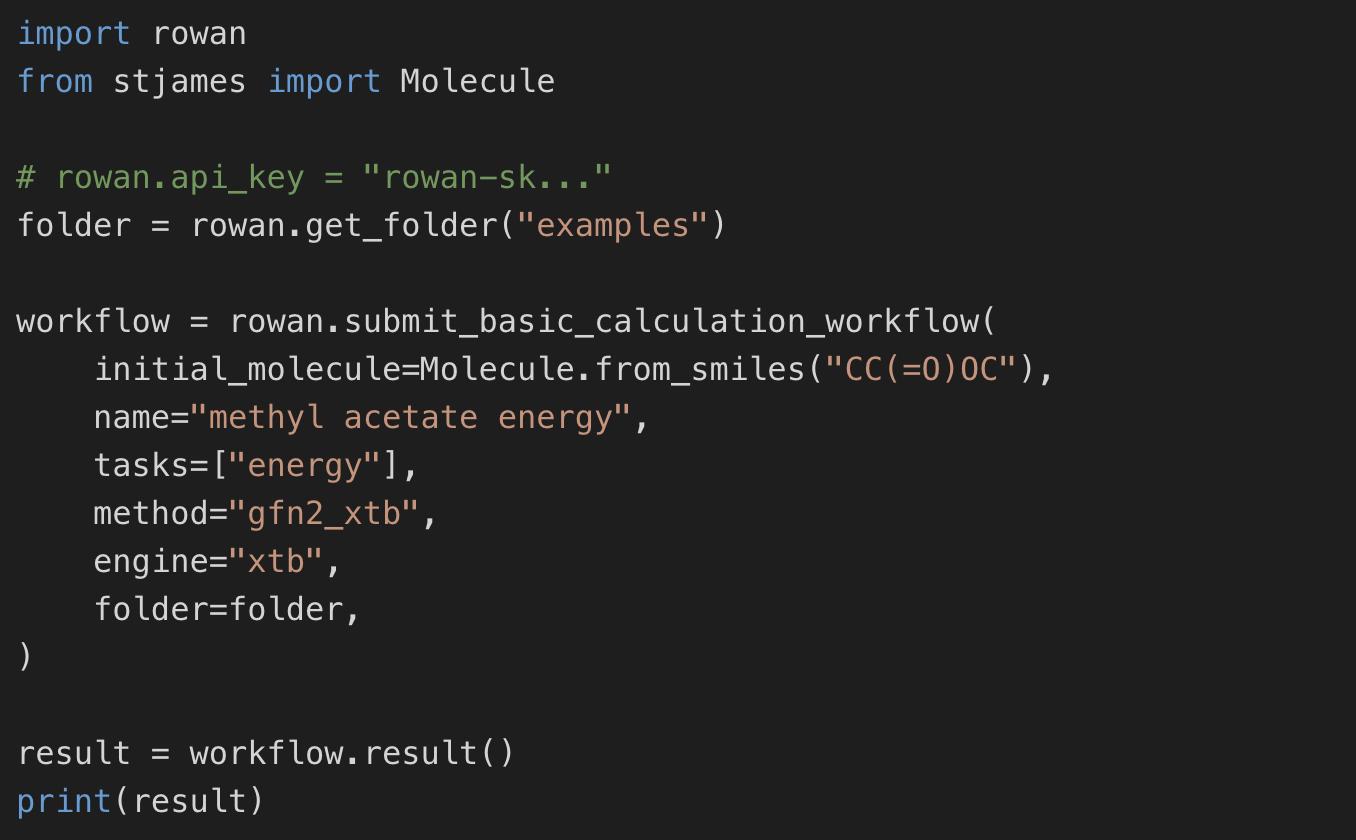
Integrate Rowan into existing computational pipelines with our Python API, or use it as a programmable backend for agentic scientific workflows.
Learn more →Data is protected with industry-standard security practices, including optional deployment into your virtual private cloud.
Learn more →Trusted by
14K+14,000+
scientists
Over
2.5M2.5 million
calculations run
Move from large-scale screening to candidate selection in one computational environment.
Hear what scientists have to say.
Rowan makes it easy for us to run reliable computational workflows without building or maintaining our own compute infrastructure. This helps us move faster across Curie's portfolio companies while keeping the focus on advancing the pipeline. The new solvent-dependent conformer workflow is an exciting, impactful addition.

Create an account to get started today or contact us to find a solution for your business.