Modern simulation tools for materials R&D
Rowan helps scientists study molecular and periodic systems with DFT, semiempirical quantum chemistry, and neural network potentials in one modern computational platform.
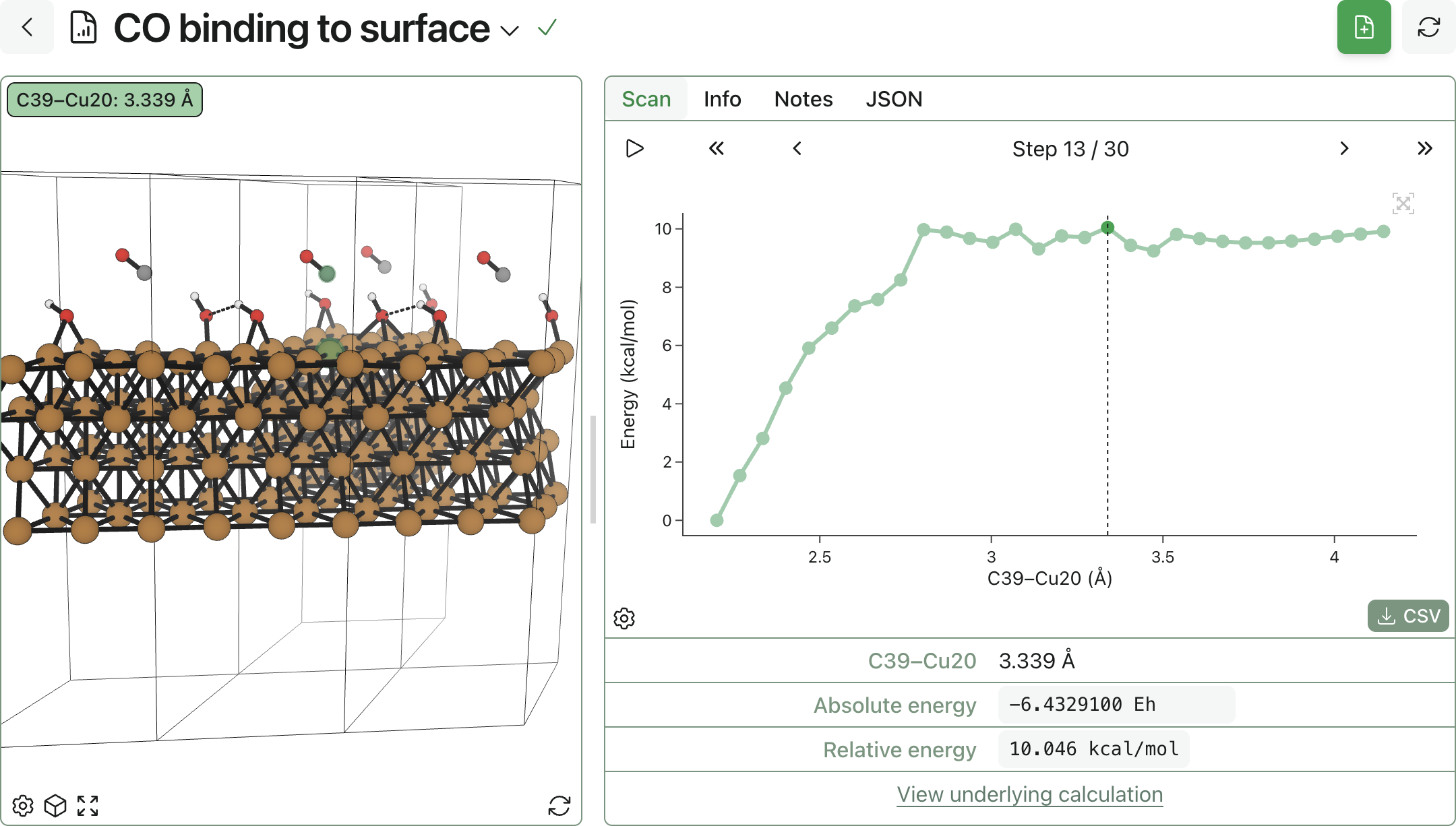
Rowan helps scientists study molecular and periodic systems with DFT, semiempirical quantum chemistry, and neural network potentials in one modern computational platform.
From catalysts and organometallic systems to molecular materials and periodic models, Rowan helps teams run practical simulation workflows without stitching together a fragile stack of disconnected tools.
Rowan helps materials teams answer questions like:
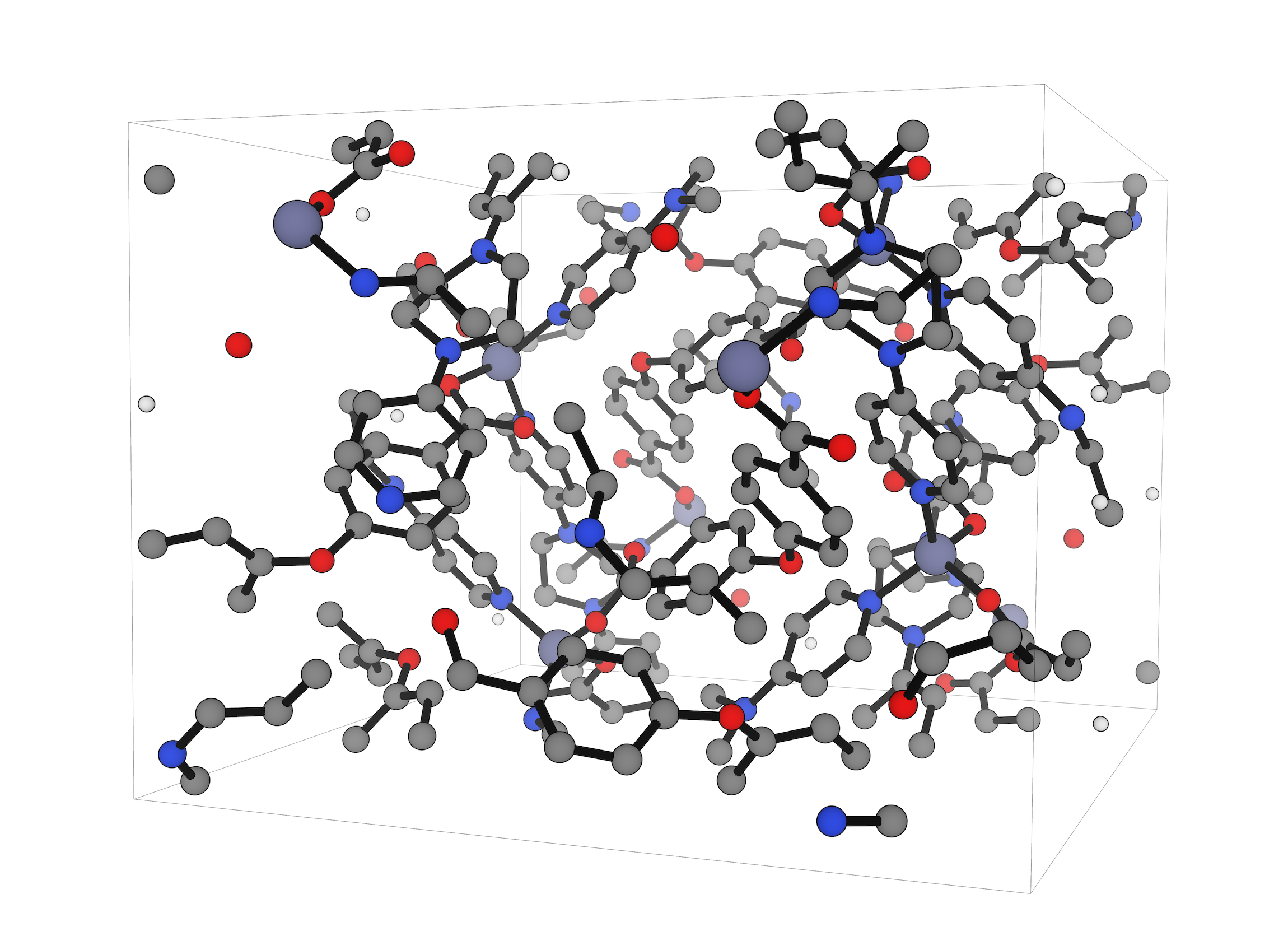
Study both discrete molecules and extended periodic systems in the same platform, from organometallic complexes and solvated molecules to solid-state and surface models.
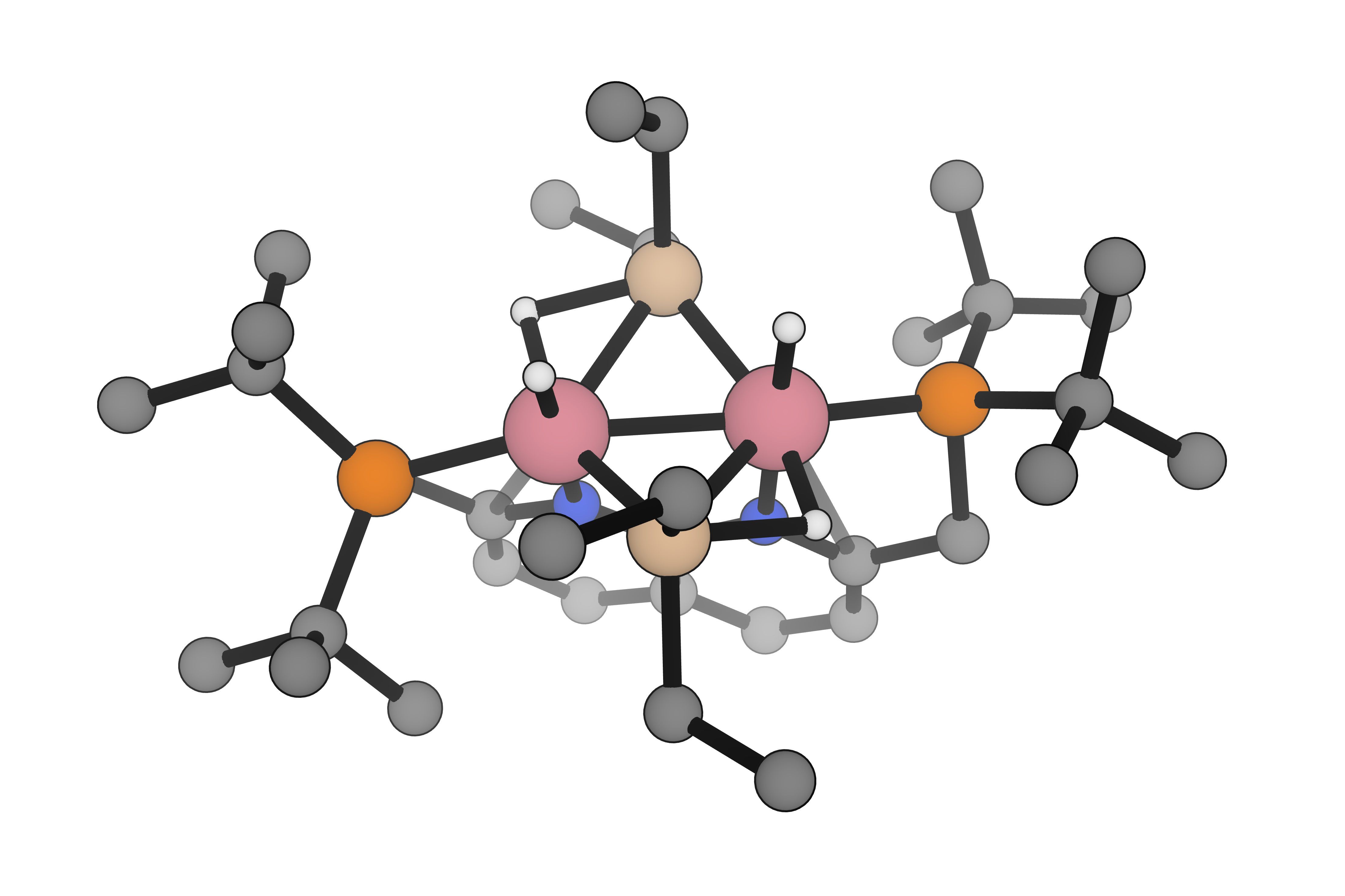
Choose the right level of theory for the problem with DFT, semiempirical quantum chemistry, and neural network potentials for screening, mechanistic studies, and large-scale simulation campaigns.
Learn more →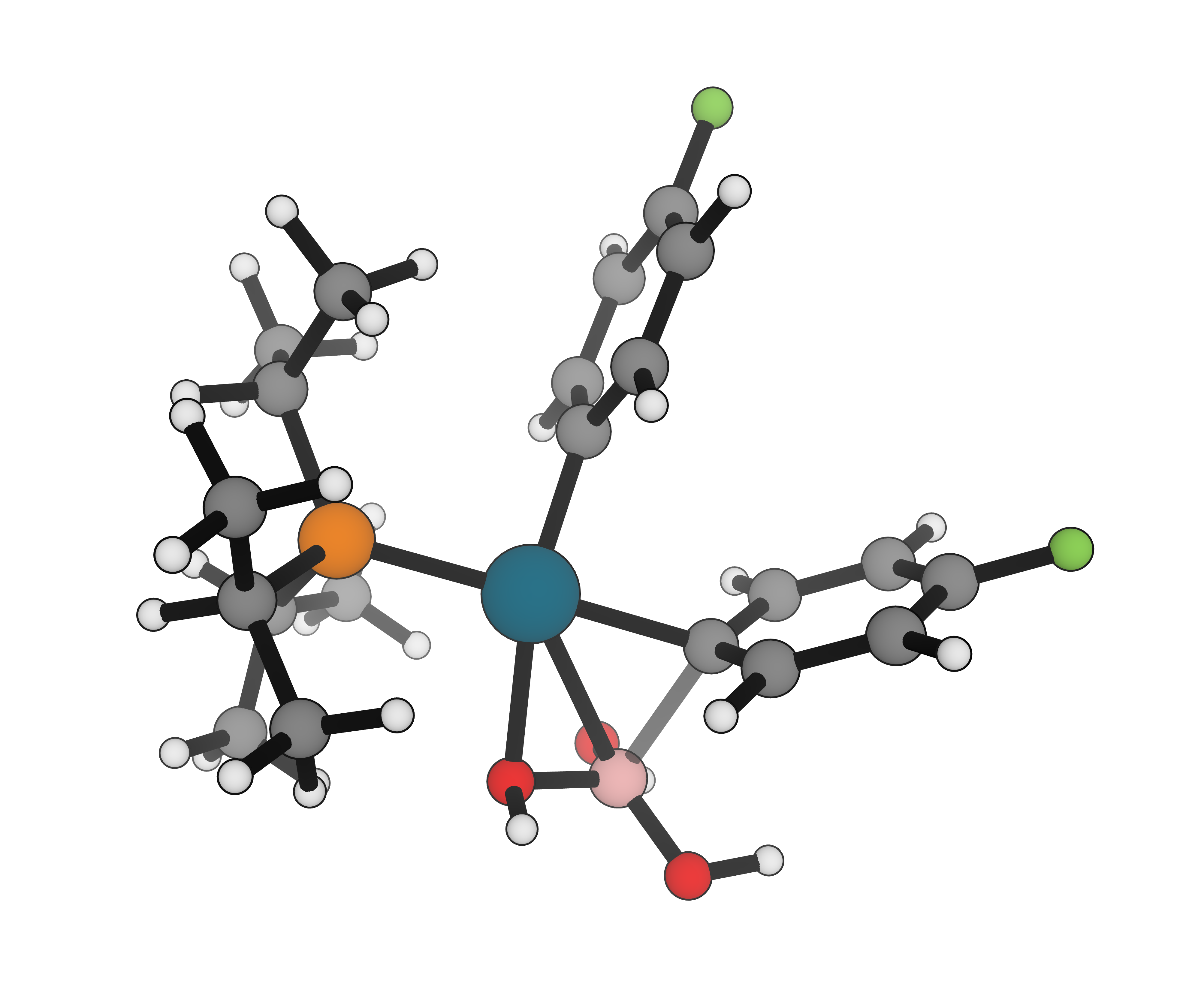
Model catalytic cycles, organometallic intermediates, spin states, and reactive species with tools designed for chemically rich systems.
Learn more →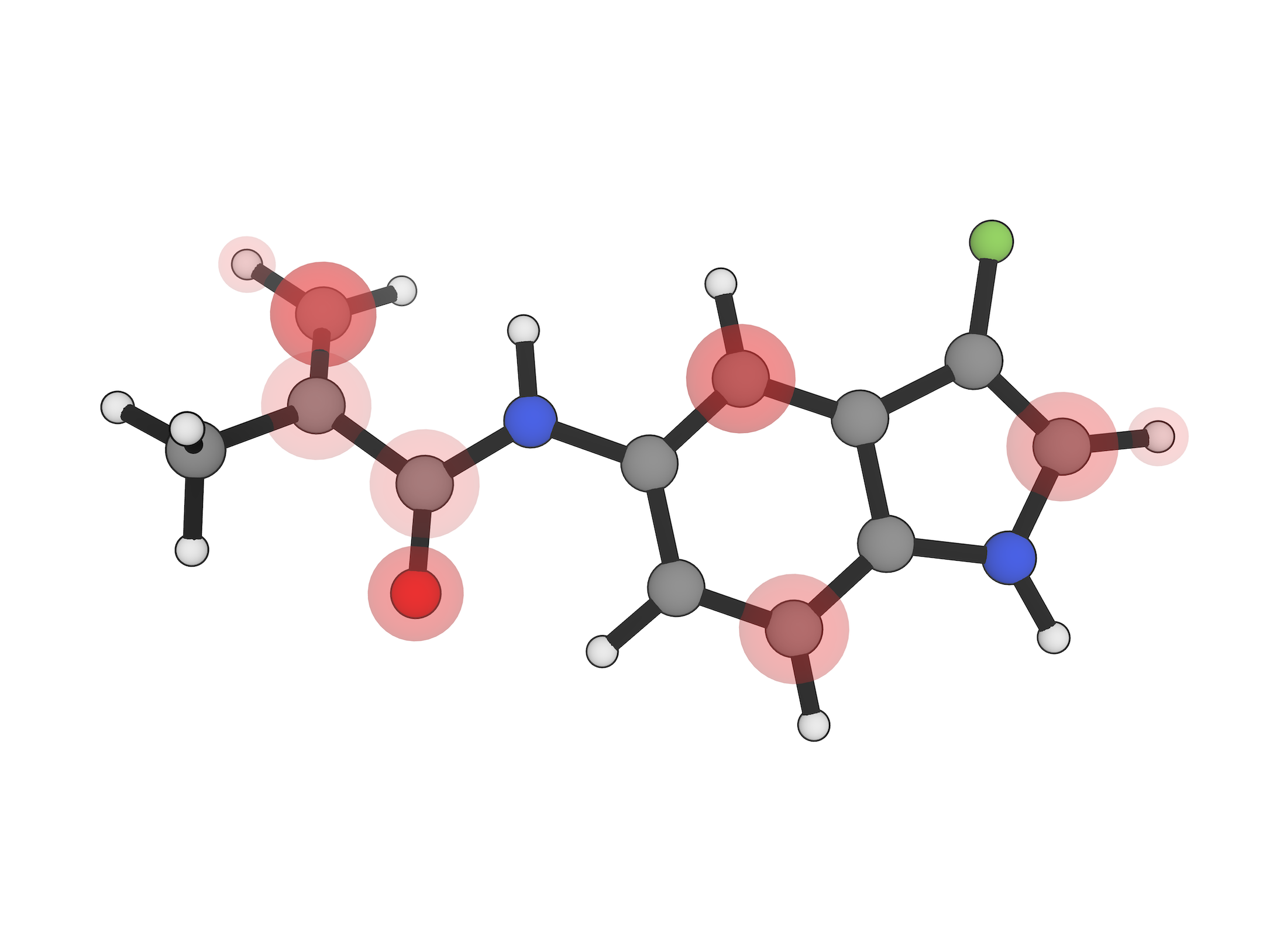
Compute Fukui indices, redox potentials, bond dissociation energies, orbitals, and other QM descriptors to understand reactivity, stability, and mechanism.
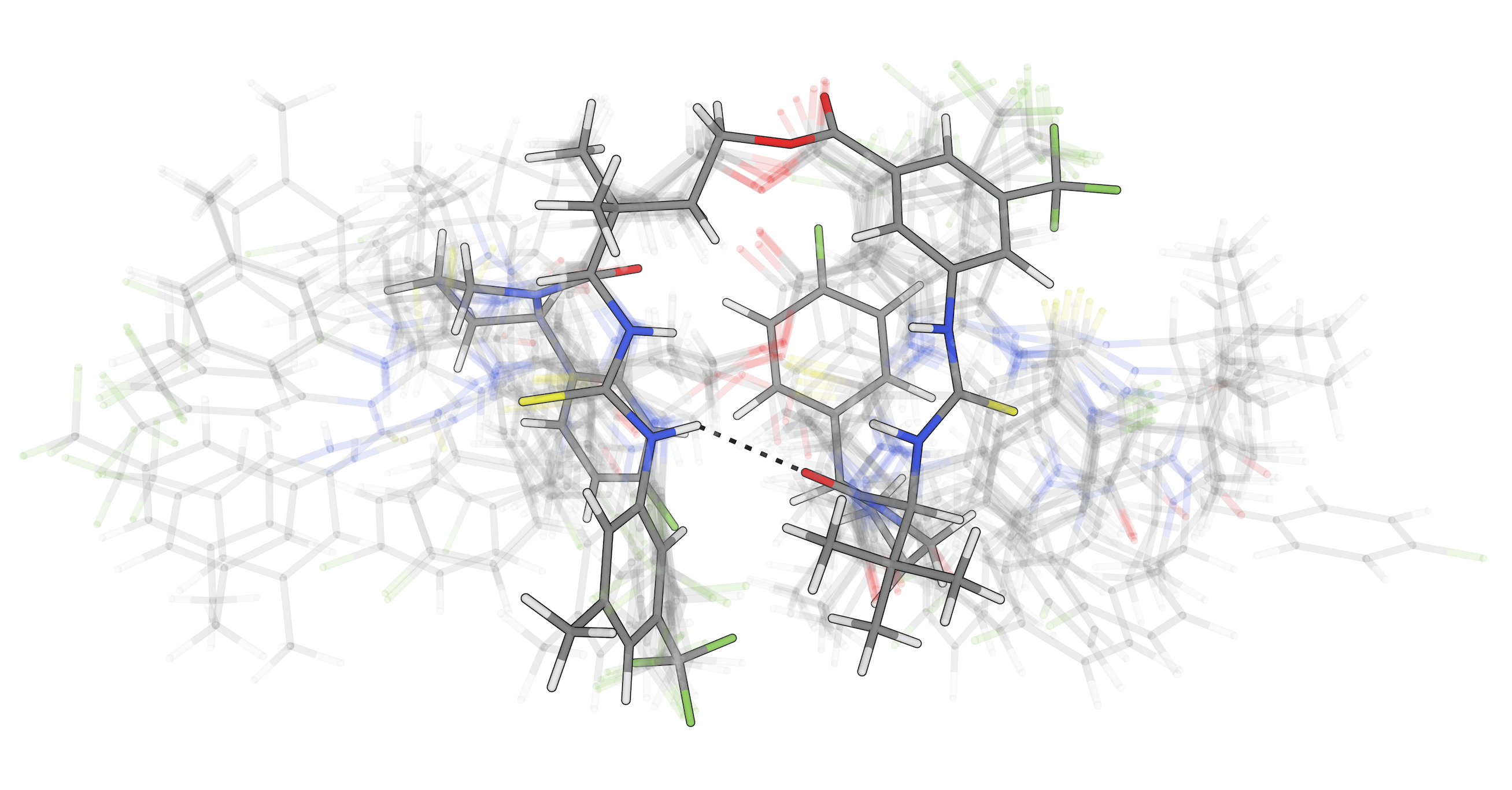
Predict solvent-dependent behavior, including conformer populations and solubility in organic solvents, to connect simulation with experimentally relevant conditions.
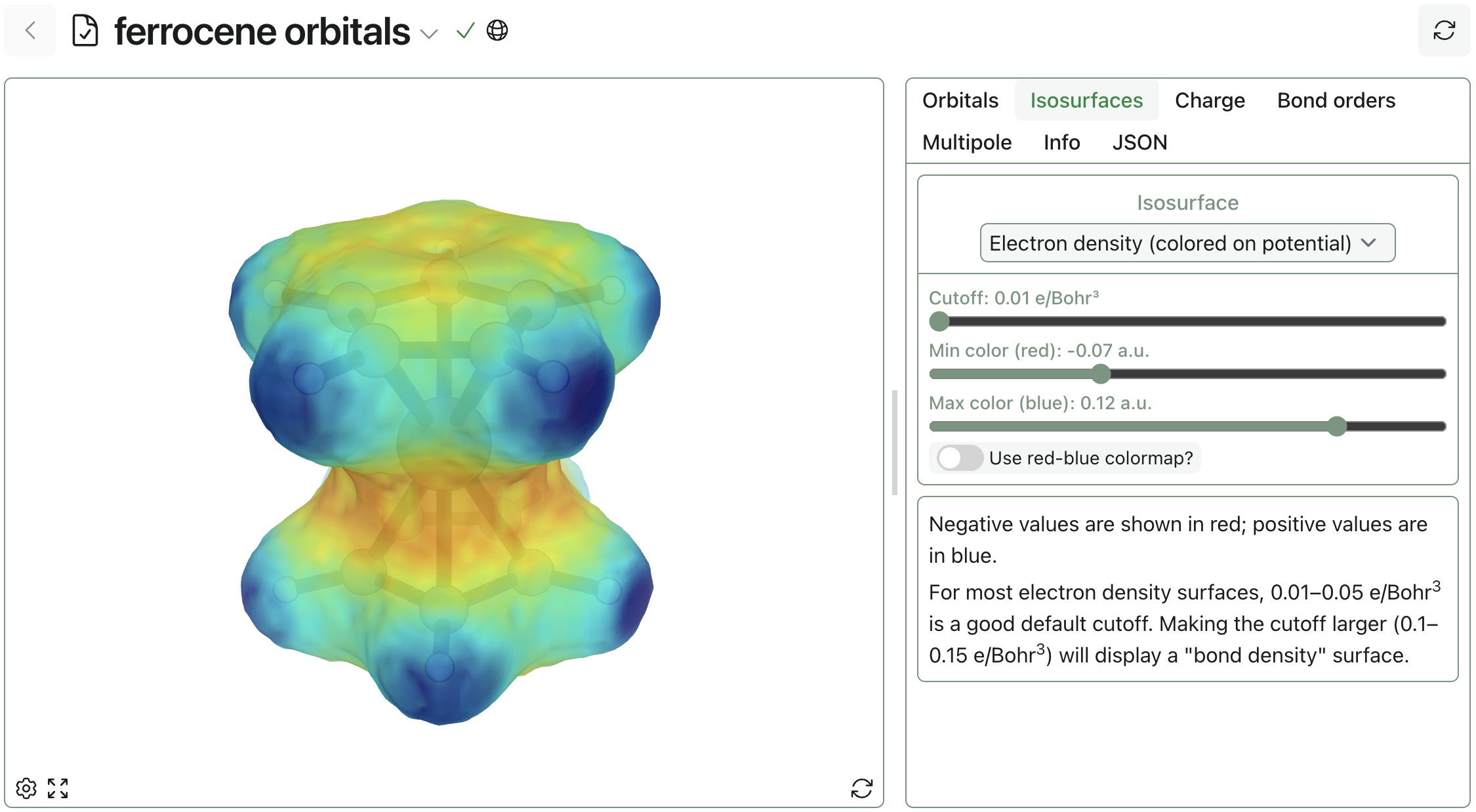
Inspect structures, orbitals, electronic properties, and computed trajectories directly in the browser, then share results securely with your team.
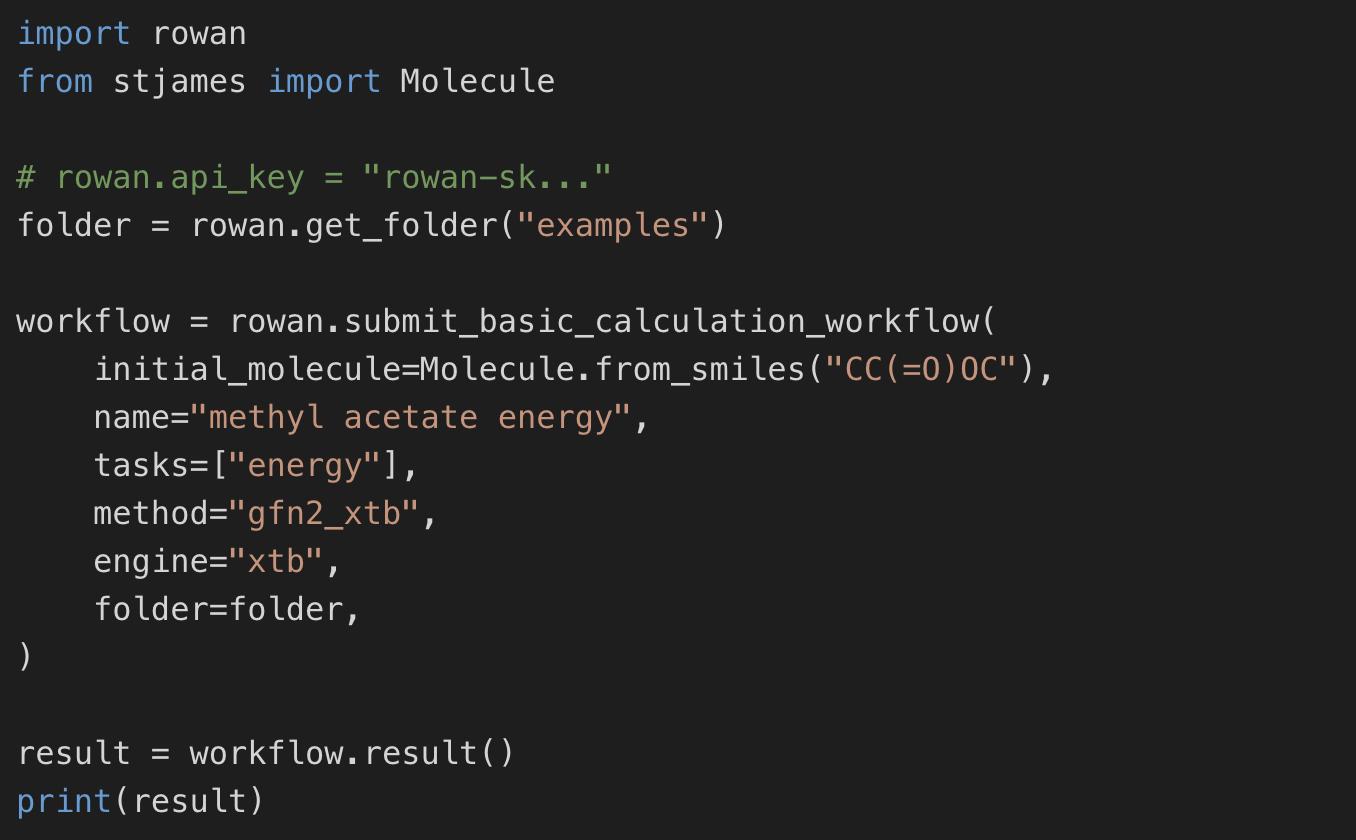
Automate screening studies, descriptor generation, and simulation workflows with Rowan's Python API.
Learn more →Protect proprietary structures, methods, and results with industry-standard security practices, including optional deployment into your virtual private cloud.
Learn more →Trusted by
14K+14,000+
scientists
Over
2.5M2.5 million
calculations run
Hear what scientists have to say.
Rowan has opened new research directions for our group. Comparing collisional cross sections calculated in Rowan versus those measured by ion-mobility mass spectrometry, we can assign structures to mixtures of diastereomers or regioisomers in an MS experiment that takes seconds.

Create an account to get started today or contact us to find a solution for your business.