Macroscopic pKa Prediction
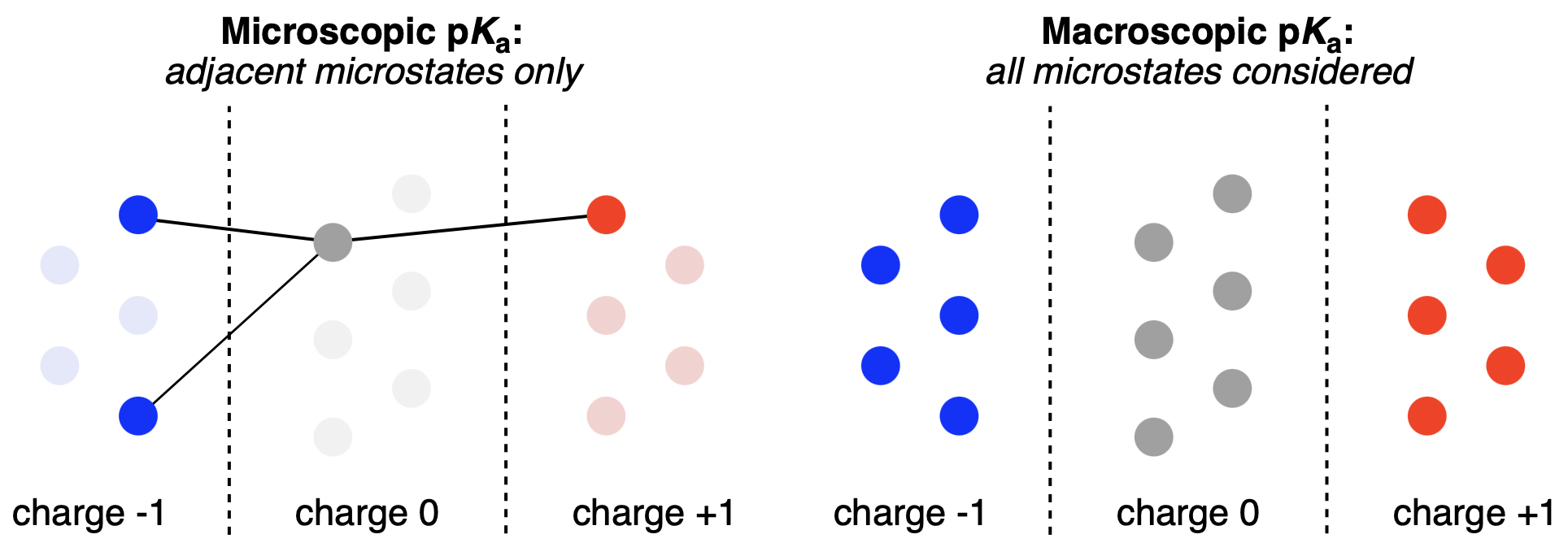
Macroscopic and microscopic pKa prediction strategies compared. Individual circles represent microstates. Figure from our macroscopic pKa work.
Most molecules of practical interest have several places that can gain or lose a proton: in water, these molecules do not exist as a single structure but as an ensemble of protonation microstates. While experimental measurements and phenomena depend on macroscopic behavior—how the average population responds to changing pH—most pKa-prediction solutions look only at a single microstate, resulting in much lower accuracy. Rowan's macro-pKa prediction workflow is built to reproduce the complexity of full macroscopic ensembles, while using machine learning to accelerate these complex calculations and make them fast enough for routine work.
(For more on the difference between the macroscopic and microscopic pKa paradigms, see our blog post.)
How Rowan's Macroscopic pKa Workflow Works
Rowan's macro-pKa workflow begins by enumerating chemically reasonable protonation and tautomeric microstates within a user-specified charge window. Chemistry-aware rules avoid implausible structures, and a beam-search strategy prevents this from becoming exponentially slower for large molecules. A compact, physics-informed neural network ("Starling") then predicts a dimensionless free energy for each microstate.
Those energies generate a partition function that produces macroscopic pKa values and, in the same pass, the population of every microstate as a function of pH. Because all quantities are derived from a single energy landscape, the output respects cycle constraints for coupled protonation and tautomerism—constraints that site-by-site methods often violate (as documented by Zheng and co-workers).
For a more detailed explanation of our methodology, see our publication.
Why It Matters
Where multiple sites or tautomers interact, the advantages of macroscopic pKa prediction become obvious. Glycine is a simple example: the canonical "neutral" microstate is not the dominant species in water, and predicting pKa from this microstate yields microscopic pKa numbers that disagree with experiment and even swap the roles of the acidic and basic groups. In contrast, Rowan's holistic ensemble-based approach reproduces the observed macroscopic values correctly.
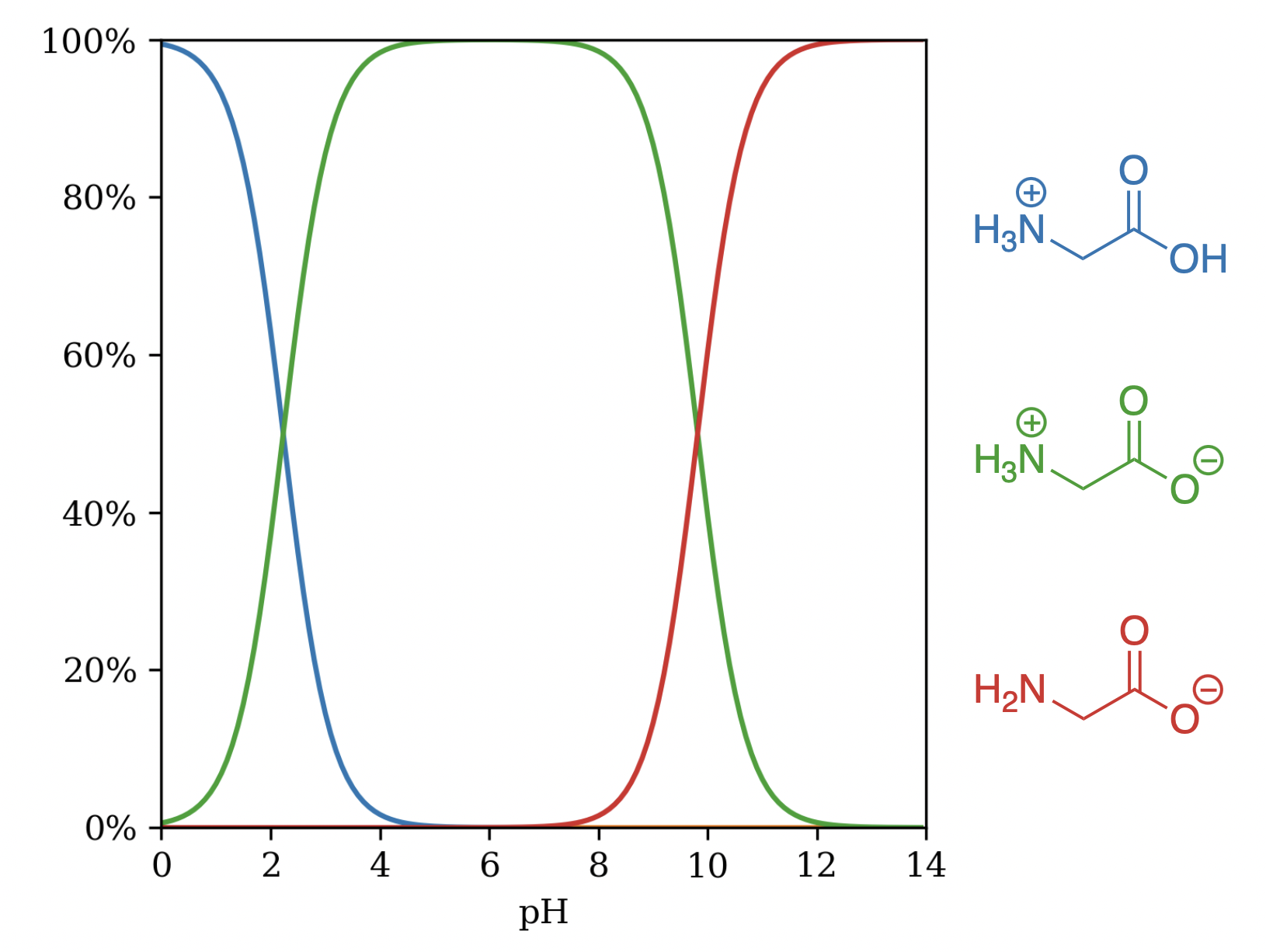
Microstates of glycine by pH.
Piperazine, a symmetric diamine, offers a second illustration. A site-by-site approach tends to predict two identical pKa values near 9, while experiment shows one near 9 and another around 5. The macro-pKa calculation recovers this separation: the first protonation alters the environment for the second and the model accounts for that shift automatically. (See the corresponding micro- and macro-pKa calculations on Rowan.)
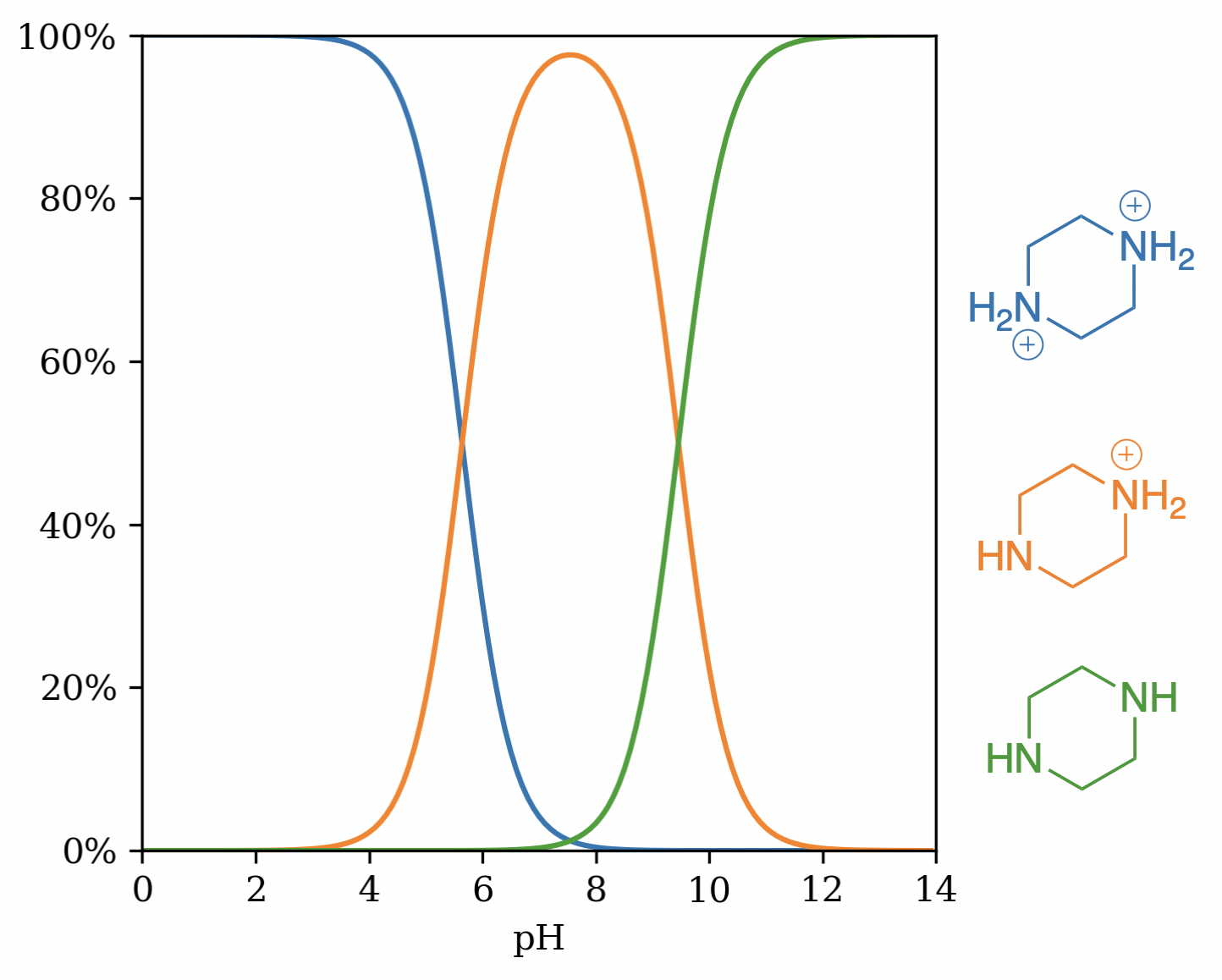
Microstates of piperazine by pH.
Where Macroscopic pKa Prediction Can Be Used
Rowan's macroscopic pKa-prediction tool can be used anywhere that pKa prediction for complex molecules is needed: when predicting charge state before a molecular-dynamics run or FEP calculation, when assessing membrane permeability, or simply in trying to understand how different substitution might affect stability or intermolecular interactions. On a variety of popular pKa-prediction benchmarks, Rowan's tool gives state-of-the-art accuracy, particularly for compounds in the biologically crucial 5–9 pKa regime.
Additionally, many properties used in pharmacokinetics and formulation depend on both intrinsic molecular traits and pH-dependent speciation. With pH-dependent microstate populations in hand, Rowan's tool can generate logD–pH profiles by combining speciation with a logP model, clarifying why a scaffold is hydrophilic in the stomach but more lipophilic in blood. For brain penetration, it provides a principled state penalty: the free-energy cost of reaching a low-charge microstate near pH 7.4. Combining that penalty with a descriptor of neutral solvation yields a simple, effective classifier for blood–brain barrier triage. The same ensemble framework can inform salt selection, crystallization conditions, permeability modeling, and protein binding—any task where the balance of neutral and charged forms matters.
How to Run Macro-pKa Calculations Through Rowan
Rowan's streamlined web-based interface makes running macroscopic-pKa predictions simple. Input the molecule or molecules you want via any of our supported file formats, select the charge states you want to study, and submit the jobs—Rowan will automatically allocate computational resources, run the calculations, and return clear publication-quality results.
For high-throughput usage, calculations can also be run through Rowan's Python API. Rowan's API is free for all users and allows hundreds or thousands of macro-pKa calculations to be submitted and analyzed programmatically. Here's what submitting a macro-pKa calculation through Rowan's API looks like:
import rowan
rowan.api_key = "rowan-sk-your-key-here"
smiles = "c1ncncc1F"
workflow = rowan.submit_macropka_workflow(
smiles,
min_pH=0,
max_pH=14,
min_charge=-2,
max_charge=2,
name=f"Macropka Workflow for {smiles}",
)
workflow.wait_for_result()
workflow.fetch_latest(in_place=True)
print(workflow.data)
print(f"View the 3D structure on the web at labs.rowansci.com/macropka/{workflow.uuid}")
If you're interested in trying out Rowan's macrocopic pKa predictions, subscribe to Rowan and start running calculations in minutes!
