How to Run Boltz-2
by Corin Wagen · Jun 6, 2025
Today, a team of researchers from MIT and Recursion released Boltz-2, an open-source protein–ligand co-folding model. Boltz-2 can not only predict the structure of biomolecular complexes from sequences, it also "approaches the accuracy of FEP-based methods" at protein–ligand binding-affinity prediction (source).
In a set of binding-affinity benchmarks, the authors show that Boltz-2 performs almost as well as the industry-standard free-energy perturbation workflow and handily outperforms cheaper physics-based methods like MM/PBSA, although performance is considerably worse on internal targets from Recursion:
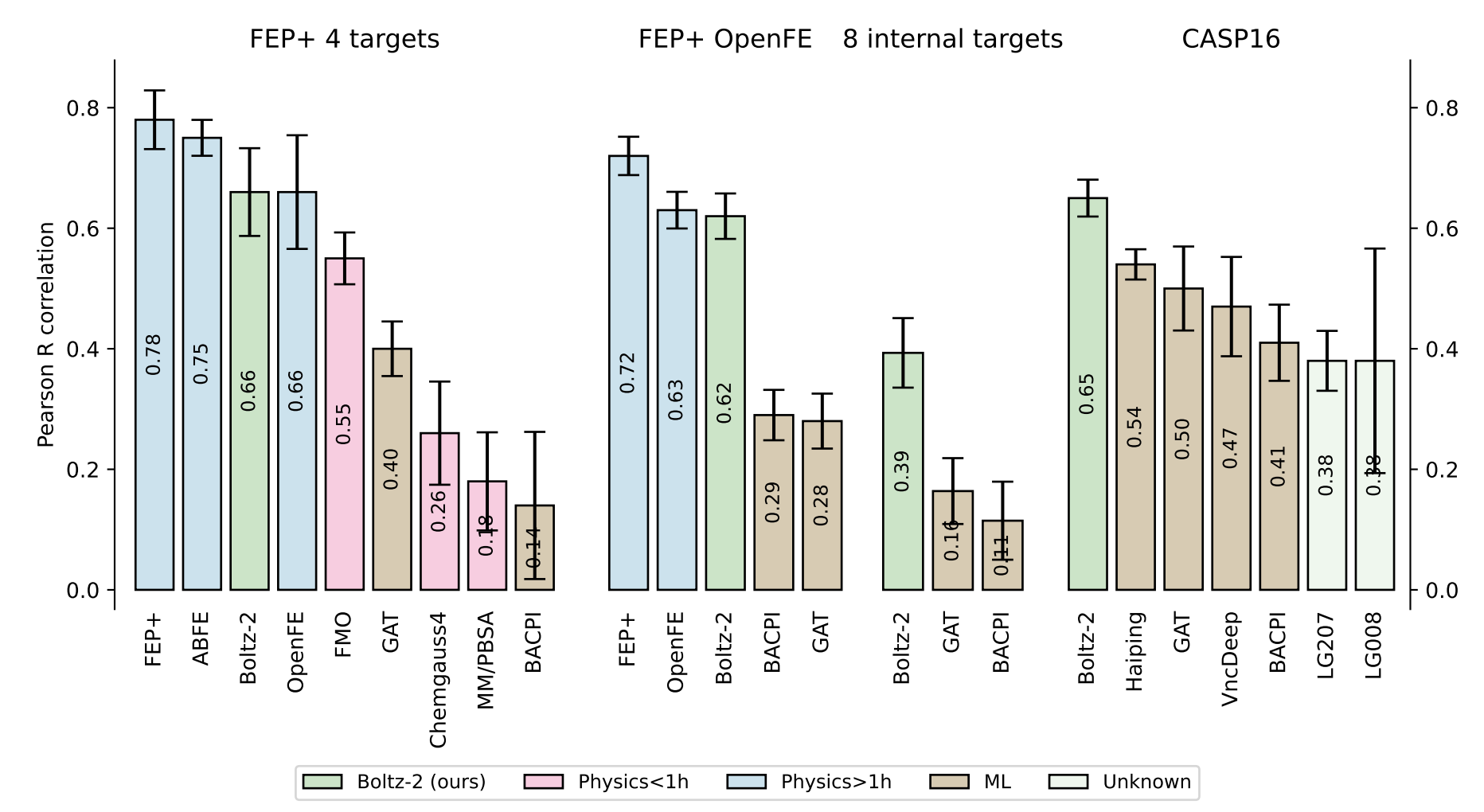
Figure 6 from the Boltz-2 paper.
While full assessment of Boltz-2's capabilities will require extensive benchmarking and external verification, it's already possible for scientists to start using Boltz-2 for their own projects. In this post, we provide step-by-step guides on how to run Boltz-2 locally and through Rowan's computational-chemistry platform.
(Curious about how Boltz-2 works? Check out this FAQ to learn more about what the model's trained on, where it can be useful, and where it still has limitations.)
Running Locally
1. Install Boltz-2
Boltz-2 is an open-source model and can be installed from the Python Package Index. You can install this any number of ways; we like using pixi for dependency management.
pixi init
pixi add python=3.12
pixi add --pypi boltz
2. Create a Template .yaml File
Boltz-2 requires a specific input-file syntax. The authors provide several examples in their GitHub repository; here's the example .yaml file for predicting protein–ligand binding affinity.
version: 1 # Optional, defaults to 1
sequences:
- protein:
id: A
sequence: MVTPEGNVSLVDESLLVGVTDEDRAVRSAHQFYERLIGLWAPAVMEAAHELGVFAALAEAPADSGELARRLDCDARAMRVLLDALYAYDVIDRIHDTNGFRYLLSAEARECLLPGTLFSLVGKFMHDINVAWPAWRNLAEVVRHGARDTSGAESPNGIAQEDYESLVGGINFWAPPIVTTLSRKLRASGRSGDATASVLDVGCGTGLYSQLLLREFPRWTATGLDVERIATLANAQALRLGVEERFATRAGDFWRGGWGTGYDLVLFANIFHLQTPASAVRLMRHAAACLAPDGLVAVVDQIVDADREPKTPQDRFALLFAASMTNTGGGDAYTFQEYEEWFTAAGLQRIETLDTPMHRILLARRATEPSAVPEGQASENLYFQ
- ligand:
id: B
smiles: 'N[C@@H](Cc1ccc(O)cc1)C(=O)O'
properties:
- affinity:
binder: B
Boltz-2 does not yet support protein–protein binding affinity or predicting binding affinity for multiple ligands.
3. Run Boltz-2 Locally
To run Boltz-2, initialize the environment shell and then use boltz predict to run the model.
pixi shell
boltz predict affinity.yaml --use_msa_server
This call will take a little while to run; make sure your computer has enough disk space to download the model weights! When finished, Boltz-2 will write a bunch of directories and .json files containing predictions. The predictions will be located in output/predictions/[input-file]/affinity-[input-file].json, and will contain predicted IC50 values (in micromolar) and binary probability that the compound is a binder.
Boltz-2 is a complex package and this guide barely scratches the surface. For a full guide to running prediction with Boltz-2, see the authors' documentation.
The latest Boltz-2 model, Boltz-2.1, is closed-source and cannot be run following the above steps. Instead, all co-folding and binding-affinity calculations must be run through the Boltz API, including workflows run by software platforms like Rowan.
Running Through Rowan
To quickly use Boltz-2 for binding-affinity prediction, calculations can also be run through Rowan. Creating an account on Rowan is completely free and can be done using any Google-managed email account; create an account here.
1. Choose Workflow
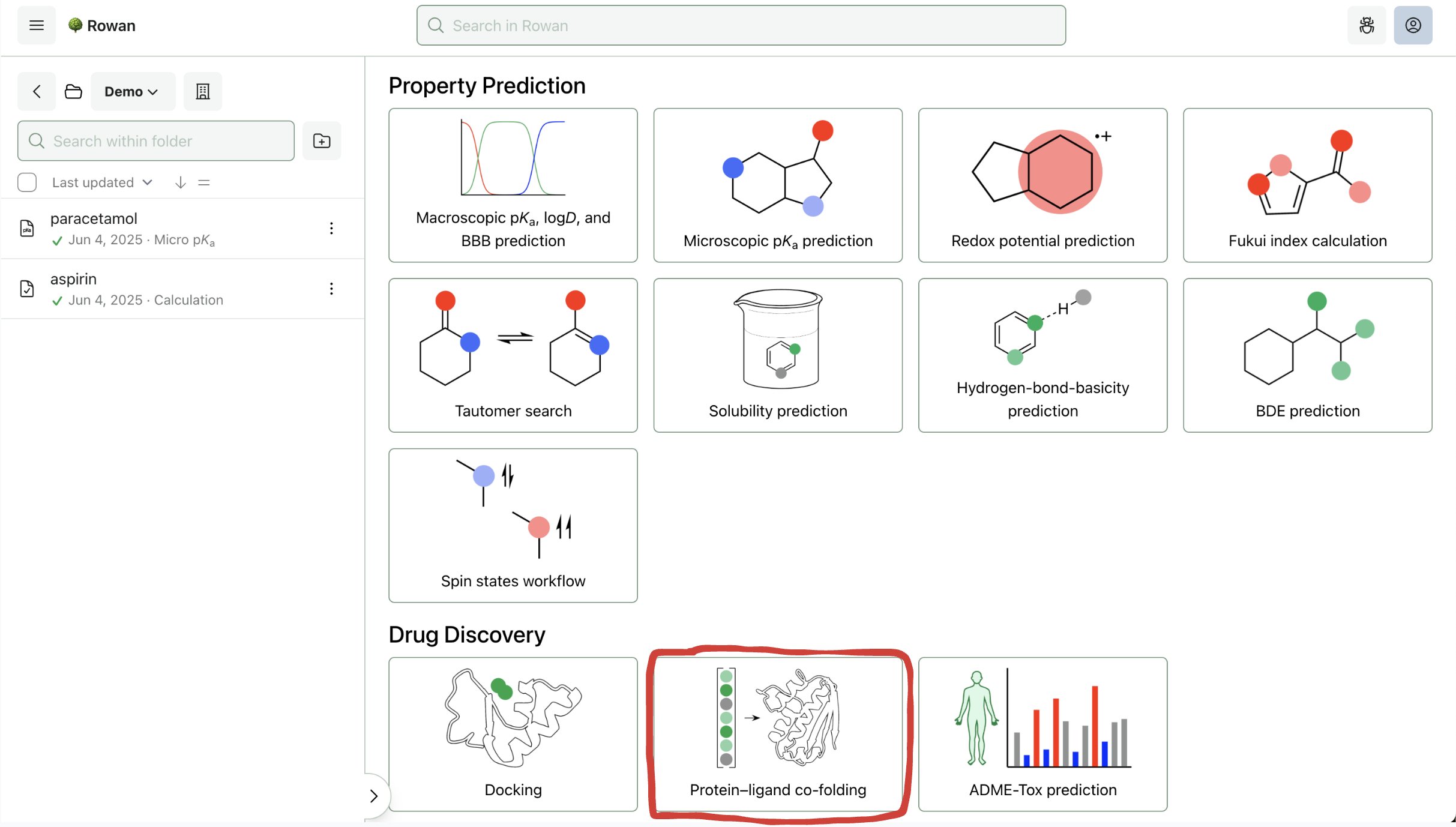
Once you sign in to Rowan, you can select which workflow you want to run. Here, we'll select the "Protein–Ligand Co-Folding" workflow (towards the bottom of the screen).
2. Enter Protein and Ligand
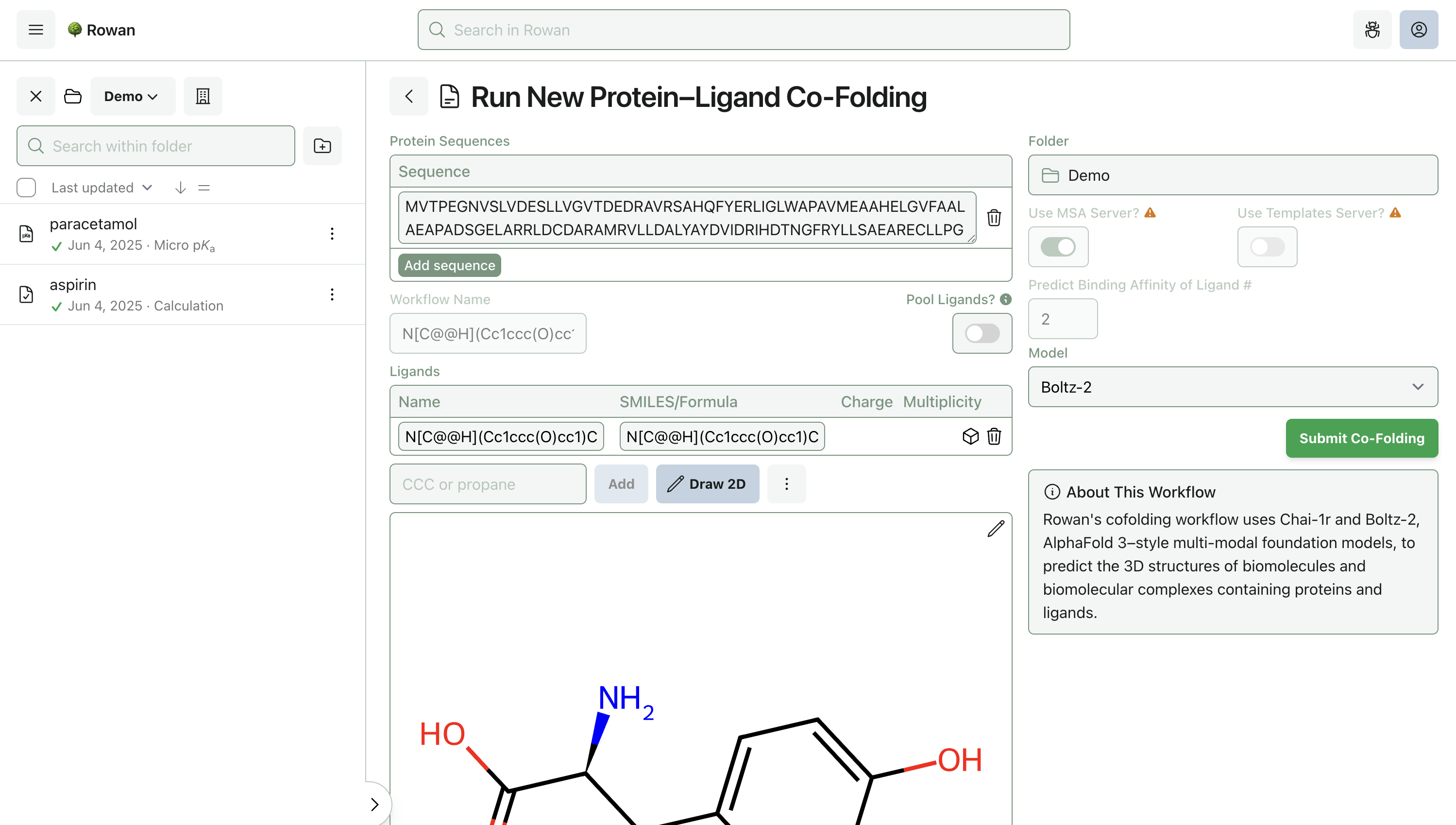
Proteins can be specified by sequence; existing protein structures in Rowan won't work, because this is co-folding—we don't want to start with a 3D structure.
Molecules can be loaded into Rowan by name, by SMILES, by input file, or through our provided 2D and 3D editors. Here, we'll input the molecule from the above demo by SMILES.
To run other co-folding models, like Boltz-2.1 or OpenFold3, change the model from "Boltz-2" to any of Rowan's other supported models.
3. Run Calculation
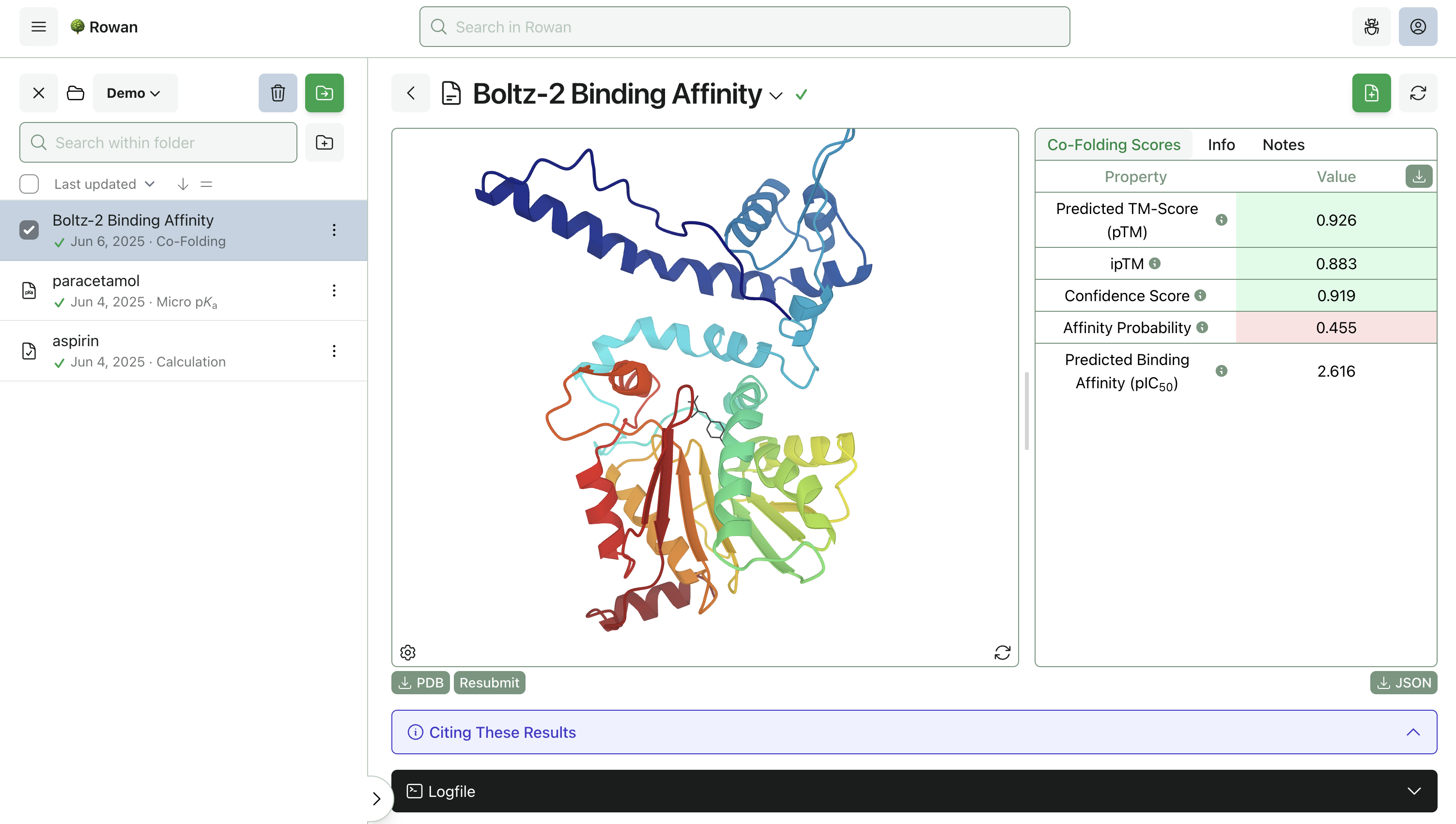
Once you click "Submit Calculation," we'll allocate a cloud GPU and start running Boltz-2 on your system. The calculations should be done in a few minutes and can be viewed through the browser.
Rowan displays the predicted protein–ligand complex through our 3D viewer, with predicted binding affinity and confidence metrics on the side. The complex can be downloaded as a PDB file for further analysis.










