Screening Conformer Ensembles with PRISM Pruner
by Nicolò Tampellini · Nov 25, 2025
This is a guest post by Nicolò Tampellini, the author of the PRISM Pruner conformer screening package we use here at Rowan. Nicolò is currently a Ph.D. student in Scott Miller's lab at Yale University, and has worked on the computational modeling of multiple conformationally complex reactions in the context of asymmetric catalysis.
Many properties in computational chemistry are obtained from conformational ensembles: sets of many spatial arrangements of the same molecule (or aggregate) that are processed as a whole to accurately model the desired property. Working with ensembles is essential when targeting the lowest energy conformations (which often influence reactivity), calculating conformational entropies, or modeling any observable property that is modulated by conformations, e.g. the shielding tensors from which to obtain NMR chemical shifts.
In many instances, the generation of such ensembles and their refinement occur in separate steps and with different levels of theory. For example, a conformational search might be carried out with an inexpensive force field or semiempirical method, but further refinement of the ensemble needs to be carried out with high-level DFT to achieve chemical accuracy. In these multi-level workflows, it often happens that multiple geometries converge to the same local minima, and a pruning step is necessary to remove duplicates and make sure to only carry forward the minimal number of structures to keep the computational cost as low as possible.
The most well-known and used metric to compare conformations is the root-mean-squared deviation (RMSD) of atomic positions. This strategy often works well, but there are some tricky caveats: different rotamers of the same structure will have artificially high RMSD values, while being chemically identical! A more ingenious solution is to compare the moments of inertia along the principal axes. This strategy is indexing-invariant, and therefore should circumvent the degenerate rotamers issue, on top of also being faster to compute.
The popular conformational search engine CREST features an ensemble sorting routine which implements both of these metrics called CREGEN. While possible to use as a standalone program, it focuses on the removal of duplicate structures while retaining all rotamers. This is necessary for some tasks like the calculation of conformational entropy, but can enormously inflate the size of ensembles if you are not interested in them. Imagine modeling some organocatalyst with a dozen tert-butyl groups! CREGEN is also written in Fortran, which can lead to difficulties when integrating into existing Python pipelines.
Born out of necessity after working with large conformational ensembles, years ago I started writing a conformational pruning implementation in Python (initially as part of FIRECODE, a modular ensemble optimization driver). Rowan indicated a need for an open-source, standalone conformer screening tool in their recent Open-Source Projects We Wish Existed blog post, and I volunteered to convert my existing code into a standalone package. Working with Jonathon from Rowan, I extracted and polished the code into PRISM Pruner.
The code implements a cached, iterative, divide-and-conquer approach on increasingly larger subsets of the ensemble and removes duplicates as assessed by the two metrics above, RMSD and moment of inertia on the principal axes. On top of that, a third mode uses a rotamer-corrected RMSD metric, in cases where the moment of inertia alone is not sufficient to weed out redundant conformations. Comparing every structure to every other requires a lot of costly evaluations, and has O(N2) scaling (where N is the number of structures). If there are a lot of similar conformers, using a divide-and-conquer strategy to group them into smaller chunks can drastically reduce the number of calls, as the small chunks keep the number of evaluations under control by using small N values as N decreases.
If energies are available, we sort the ensemble before dividing it into chunks to have the best chance of grouping similar structures together early. After all chunks are evaluated, the leftover structures are used to repeat the process with larger chunks, until all active structures are included in the final evaluation. In many instances this results in significantly fewer comparisons, and a faster and more scalable algorithm. Even in the worst case of no similar structures, the use of a cache ensures that we don't ever perform more calls than a simple all-to-all algorithm would.
Our initial comparisons against CREGEN are very positive, particularly for larger ensembles with many identical rotamers, where the divide-and-conquer approach really shines. The worst-case scaling factor is still O(N2) if all conformers are different, but functionally it is much lower for most conformational ensembles.
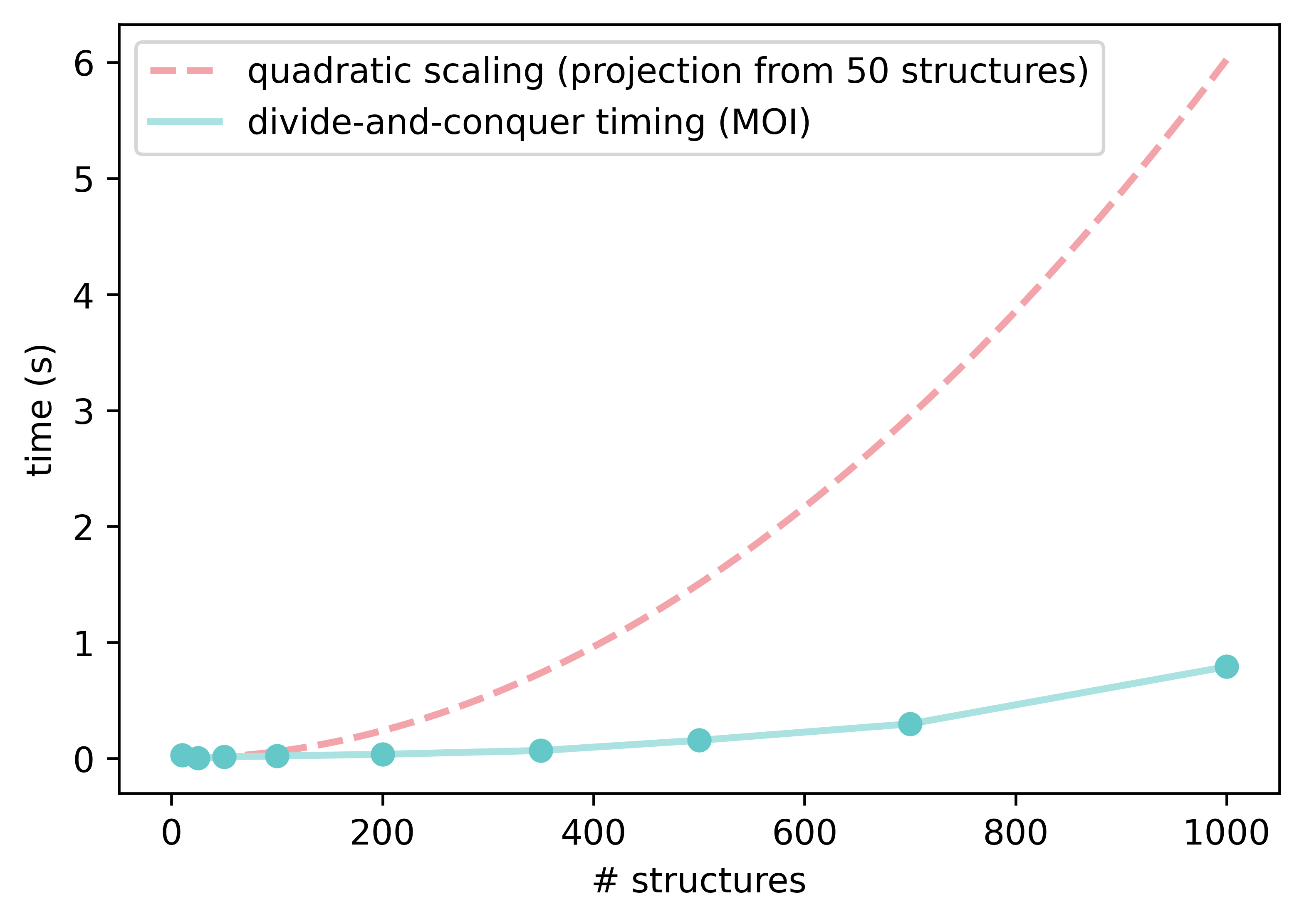
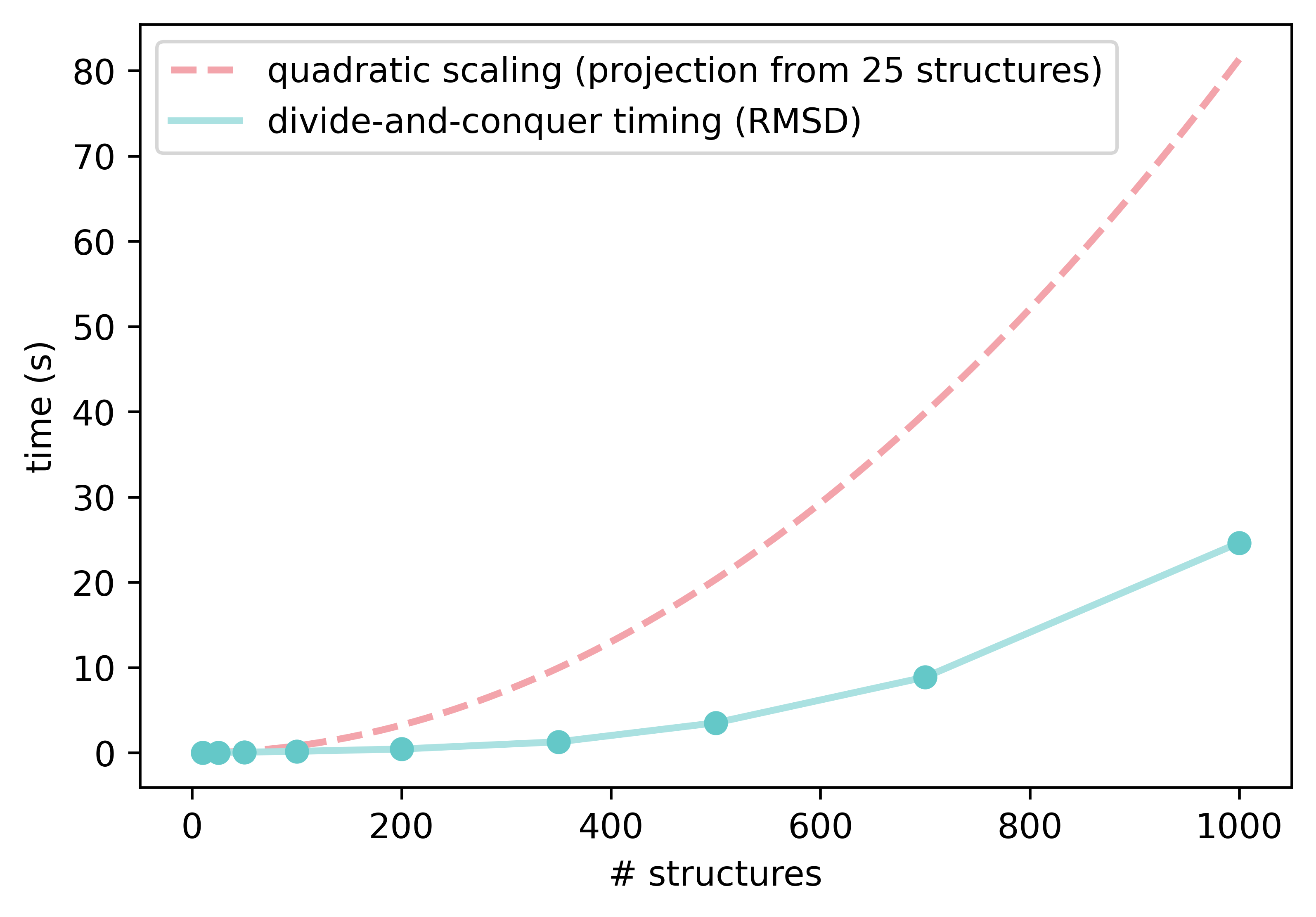
We have also added a convenience function to perform sequential pruning, using reasonable default values for each step: starting with the fast moment of inertia mode, it follows with RMSD-based pruning and then an optional, final, rotamer-corrected RMSD pruning. Processing ensembles with ≈1,000 structures of ≈150 atoms using these settings takes seconds, and removes many rotamers from ensembles obtained from CREST. Here are two examples from my Ph.D. work, showing how much a conformational ensemble can be inflated by undesired rotamers—the second one is really pathological! The DFT time saved by processing these ensembles before the next step is significant.

The future of this project is also in your hands: if you are interested in contributing with new features, feel free to reach out to me or open a request on GitHub! For example, more similarity evaluation metrics could be implemented to screen for specific conformational attributes.
The performance of some sections could also be improved, if needed: while the MOI-based evaluation of similarity is really fast, the RMSD evaluation with numpy alone could be faster. The original FIRECODE implementation of the RMSD metric relies on Numba, which compiles low-level code at runtime and achieves a ≈7x speedup on the numerically-intensive RMSD calculation. While really performant, the Numba library is very heavy, and can complicate integration into packages already containing a large number of dependencies, thus we decided not to include it. If you see further room for improvement in the code performance, we'd love to hear from you!










