The Boltz-2 FAQ
by Corin Wagen and Ari Wagen · Jun 9, 2025
Over the past few days we've had hundreds of scientists sign up for Rowan to run Boltz-2, many of whom have questions about how it works, where it's useful, and when it might fail.
To address these questions, we've put together the following practical FAQ page about Boltz-2. Enjoy!
Note: this FAQ does not cover the new BoltzGen model, although Google sometimes redirects searches about BoltzGen to this page. If you're curious about BoltzGen, see our dedicated BoltzGen page.
1. What is Boltz-2?
Boltz-2 is a multimodal "co-folding" model that simultaneously predicts 3D structures of protein, DNA, RNA and small-molecule complexes and binding affinity (both continuous values and binder/decoy likelihood) for protein–ligand pairs.
According to the paper, Boltz-2 matches or exceeds state-of-the-art structure accuracy across most modalities, and is the first AI model to approach free-energy-perturbation accuracy while being ~1000× faster in typical affinity calculations. (If you're curious what this means in practice, keep reading!)
2. How is Boltz-2 different from Boltz-1 and other models?
Boltz-2 is similar to Boltz-1, but contains numerous enhancements and new capabilities. Here's a brief overview:
| Aspect | Boltz-1 | Boltz-2 |
|---|---|---|
| Structural engine | 48 PairFormer layers, 512-token crop | 64 layers, trifast kernels, 768-token crop |
| Controllability | None | • Method conditioning (X-ray / NMR / MD) • Multi-chain template steering • Contact & pocket constraints |
| Physics quality | Optional steering potential (Boltz-1x) | Optional steering potential (Boltz-2x) |
| Affinity head | None | PairFormer-based dual head (probability + pIC50) |
| MSA | Optional | Required |
Both models can be run through the same package, allowing for easy comparison of results.
Here's a few more details on features from the above table.
2.1 Binding-affinity prediction
Boltz-2 can predict binding affinities through a new output head, which outputs both the probability that a given ligand is a binder and a quantitative estimate of the IC50 for the ligand.
Subsequent statements from the Boltz-2 team indicate that these two outputs are intended to be used in different ways. The first output, affinity_probability_binary, is intended for hit discovery and "should be used to detect binders from decoys, for example in a hit-discovery stage." In contrast, the affinity_pred_value output can be used to predict ligand SAR and "should be used in ligand optimization stages such as hit-to-lead and lead-optimization."
Ligands with 50 or more atoms are poorly represented in the Boltz-2 training dataset. While early Boltz-2 releases allowed affinity calculations to be run on large molecules, newer versions of Boltz-2 can no longer compute binding affinity for ligands with 128 atoms or more (GitHub commit).
The newest version of Boltz-2, "Boltz-2.1," contains substantial changes to the binding-affinity-prediction head which replaces the above affinity outputs with two new ones: binding_confidence and optimization_score. The above section should be presumed invalid until further benchmarking is conducted.
2.2 Method conditioning
Method conditioning allows scientists to hint Boltz-2 about the experimental context (static X-ray, solution NMR, dynamic MD, etc) so it biases its sampling toward conformations typical of that technique. Here's the current list of methods supported by Boltz-2 (source):
- MD simulations (
md) – structures or ensembles derived from classical or enhanced-sampling molecular dynamics. - X-ray diffraction (
x-ray diffraction) – atomic models solved from single-crystal X-ray data. - Electron microscopy (
electron microscopy) – cryo-EM or negative-stain reconstructions refined into atomic coordinates. - Solution NMR (
solution nmr) – conformations obtained from nuclear-magnetic-resonance restraints in solution. - Solid-state NMR (
solid-state nmr) – structures determined by NMR on crystalline, fibrous or membrane samples. - Neutron diffraction (
neutron diffraction) – crystal structures refined against neutron diffraction data (often with visible hydrogens). - Electron crystallography (
electron crystallography) – diffraction from 2-D crystals analysed by electron beams. - Fiber diffraction (
fiber diffraction) – helical or fibrous assemblies solved via X-ray fiber patterns. - Powder diffraction (
powder diffraction) – structural models derived from polycrystalline powder X-ray data. - Infrared spectroscopy (
infrared spectroscopy) – coarse structural constraints inferred from IR/FTIR spectra. - Fluorescence transfer (
fluorescence transfer) – distance information from FRET or similar fluorescence techniques. - EPR / DEER (
epr) – spin-label electron-paramagnetic-resonance measurements providing inter-spin distances. - Theoretical model (
theoretical model) – purely in-silico or energy-minimised coordinates without direct experimental data. - Solution scattering (
solution scattering) – low-resolution shapes from SAXS/SANS fitted to atomic models. - Other (
other) – any experimental or computational source not covered above. - AFDB predictions (
afdb) – coordinates taken from the AlphaFold Database. - Boltz-1 predictions (
boltz-1) – legacy structures generated by the Boltz-1 co-folding model.
When running Boltz-2 (see below), method conditioning can be specified by the --method keyword.
2.3 Multi-chain template steering
Scientists can supply one or more existing structures for specific chains and the model will keep those regions close to the template while freely folding the rest of the complex.
2.4 Contact & pocket constraints
Scientists can define residue-pair distances or a set of pocket residues. Boltz-2 will apply soft potentials that nudge the prediction to satisfy those geometric constraints.
2.5 Multiple sequence alignment (MSA)
Boltz-2 requires MSA values to give good results, although in emergencies they can be omitted. By default, Boltz-2 uses the public api.colabfold.com MSA server hosted by KOBIC, the Korean Bioinformatics Center.
Rowan hosts a private MSA server, allowing Rowan-hosted Boltz-2 jobs to run without sending data to external servers. This also prevents Rowan's Boltz-2 calculations from failing when the api.colabfold.com experiences outages (like in August 2025).
3. How do I run Boltz-2?
3.1 Command-Line Usage
Boltz-2 can be run through the open-source boltz package on GitHub. Once boltz has been installed, Boltz-2 can be run through the command-line:
boltz predict <INPUT_PATH> [OPTIONS]
There are extensive directions for all the different command-line options that can be employed to run Boltz-2. At a high level, inputs to Boltz-2 can be provided through two formats:
- YAML (recommended) – full control over chains, ligands, covalent bonds, templates, pocket constraints, custom MSAs, etc.
- FASTA – quick single-sequence or MSA-backed predictions.
Importantly, there's currently no way to run binding-affinity predictions through FASTA input, so any previous FASTA-based workflows will need to convert to YAML to incorporate binding affinity.
For a simple example script with installation directions, check out our guide to running Boltz-2. For more advanced usage, various command-line options can be modified. Here are some relevant arguments:
| Flag | Purpose | Default |
|---|---|---|
--recycling_steps | Iterative refinement passes. | 3 |
--sampling_steps | Diffusion timesteps per sample. | 200 |
--diffusion_samples | Number of independent poses per input. | 1 |
--step_scale | Controls diversity; lower means more diverse. | 1.638 |
--use_potentials | Activate physics steering (Boltz-2x). | False |
--output_format | Changes output file format; pdb also available. | mmcif |
The output of Boltz-2 is a set of nested directories following the below schema:
out_dir/
└── predictions/
└── <input>/ # one folder per input
├── *_model_0.cif # ranked structures
├── confidence_*.json
├── affinity_*.json
└── pae_/pde_/plddt_*.npz
Several points merit further discussion:
- Confidence scores are calculated as
0.8 * plddt + 0.2 * ipTM - Affinity values are given in log(IC50 µM) units, which can be converted to kcal/mol through the following expression:
(6 - affinity) * 1.364. (This is a non-standard pIC50, so use caution when comparing to predictions from other software packages.)
For a full discussion of output format, refer to the above documentation.
The new Boltz-2.1 method is closed-source and cannot be run locally; instead all inference must be run through the Boltz-2 API.
3.2 Hosted Boltz-2 Inference
Boltz-2 can also be run through computational platforms like Rowan, which automatically parse inputs & outputs and provision GPU resources for each computation. These platforms allow users without previous programming or machine-learning experience to benefit from these new computational advances.
For a step-by-step guide to running Boltz-2 on Rowan, check out our tutorial.
4. Where might Boltz-2 be useful?
Although full assessment of Boltz-2's capabilities will require independent external evaluation and benchmarking, the authors' results suggest that Boltz-2 will be useful in several contexts.
4.1 Hit discovery & virtual screening
Boltz-2 excels at picking true binders out of very unbalanced screening libraries. On the MF-PCBA benchmark it nearly doubles the mean average precision (AP ≈ 0.025) and delivers an enrichment factor (EF) of ~18 over the top 0.5 % of the ranked list, whereas docking (Chemgauss4) and other ML baselines plateau around AP ≈ 0.005 and EF ≈ 2–3.
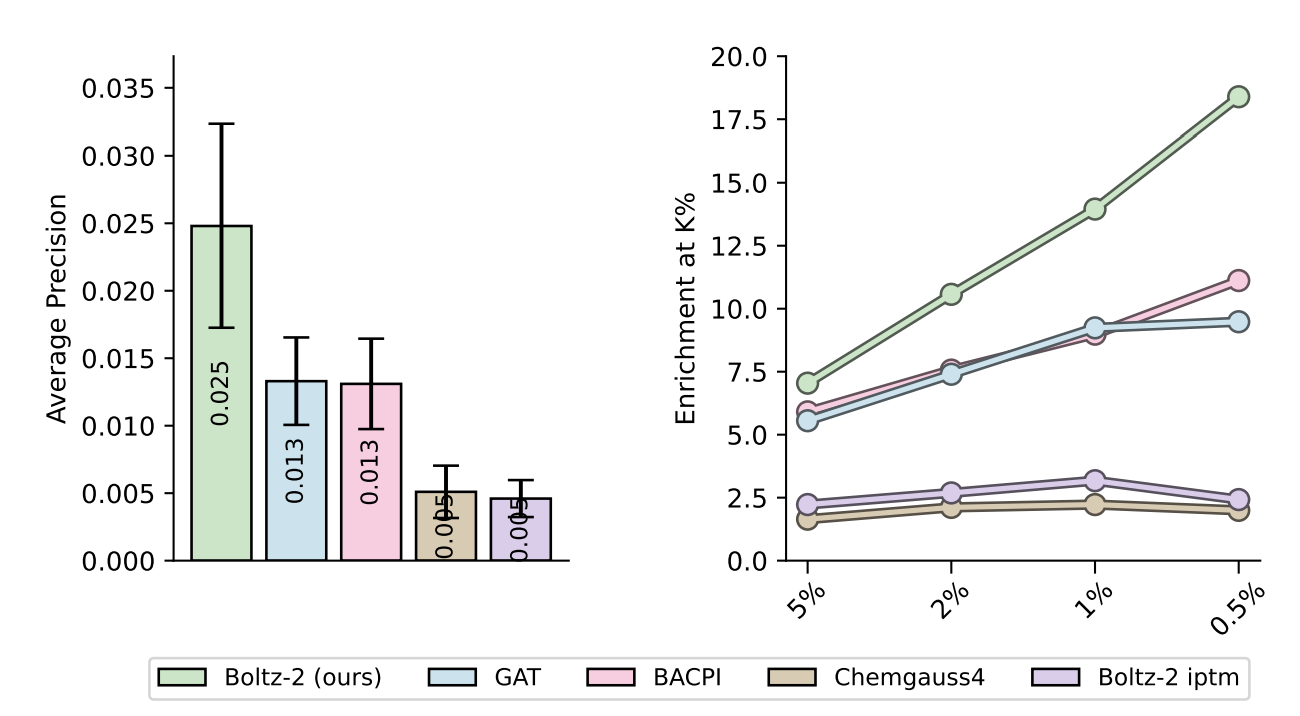
Figure 7 from the Boltz-2 paper.
Boltz-2 has a throughput of hundreds of thousands of molecules per day on an 8-GPU node, making Boltz-2 a good first-pass filter before more exhaustive docking or physics-based scoring methods.
4.2 Lead optimization
For series where subtle SAR differences matter, Boltz-2's affinity head approaches the accuracy of rigorous free-energy methods while remaining three orders of magnitude faster. On the canonical 4-target FEP+ subset (CDK2, TYK2, JNK1, p38) Boltz-2 achieves a Pearson correlation of 0.66, on par with OpenFE (0.66) and somewhat worse than commercial FEP+ (0.78).
In contrast to these methods, though, Boltz-2 requires only minutes of GPU time instead of hours or days. This makes Boltz-2 an attractive choice for rapidly prioritizing promising analogues before committing substantial resources to FEP or synthesis.
4.3 High-quality pose generation
Boltz-2's co-folded structures are docking-quality or better. In the Polaris ASAP-Discovery ligand-pose challenge (SARS-CoV-2 & MERS-CoV main proteases) the out-of-the-box model matched the top 5 finetuned competition entries and outperformed Boltz-1 without any physics-based relaxation. For antibody–antigen interfaces and other difficult modalities it likewise narrows, though does not yet eliminate, the gap to AlphaFold 3.
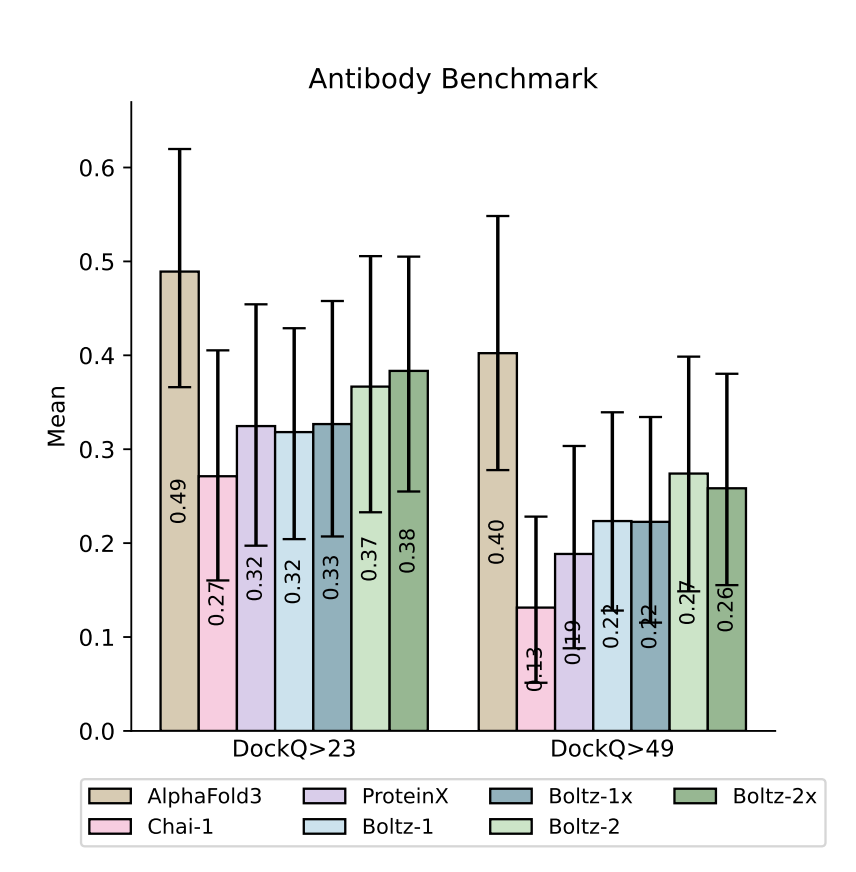
Figure 4 from the Boltz-2 paper.
4.4 Template-guided or constraint-guided modelling
Boltz-2 introduces multi-chain template conditioning plus optional steering potentials that can force portions of a prediction to stay within a chosen RMSD window of a supplied structure. It also supports residue-pair "contact", pocket and distance constraints as pairwise features with accompanying steering potentials.
5. What are the current limitations of Boltz-2?
5.1 Large conformational changes
Boltz-2 co-folds complexes from static sequence/SMILES inputs and therefore struggles with motions that occur only after ligand binding or over micro- to millisecond timescales (e.g., hinge opening, domain swaps, large allosteric shifts).
If large rearrangements are expected, considering combining Boltz-2 with molecular dynamics to gain an understanding of protein conformational motion.
5.2 Cofactors, ordered waters and ions
The current affinity head processes only a cropped set of protein and ligand heavy atoms. It does not model water-mediated bridges, metal coordination or multimeric cofactors when scoring binding; in cases where essential ions or cofactors are present in the binding pocket, affinity predictions will be unreliable.
5.3 Certain protein classes (e.g., GPCRs, ion channels)
Benchmark variance is larger for highly flexible, membrane-embedded or multistate receptors. Training data for GPCRs, transporters and ion channels is sparser than for soluble enzymes, so confidence metrics alone may not be enough to tell when Boltz-2 will give low-quality predictions.
5.4 Overall accuracy
While Boltz-2 has similar accuracy to some free-energy methods on some benchmarks, performance still lags the industry-standard FEP+ method, indicating that Boltz-2 is not yet a lossless replacement for conventional physics-based simulation.
5.5 Protein–protein binding affinity
Boltz-2 does not support computation of protein–protein binding affinity; only small molecules are supported for binding-affinity predictions.
On Github, Gabriele Corso warned that molecules with more than 50 atoms will give unreliable binding-affinity predictions.
5.6 Ligand stereochemistry and conformations
A number of scientists have reported that Boltz-2 generates incorrect stereoisomers or unrealistic conformations for small-molecule ligands. Rowan now automatically checks for this by running chirality checks, PoseBusters, and (optionally) ligand-strain calculations.
5.7 Large ligands
As mentioned above, ligands with more than 50 atoms lead to binding-affinity predictions of questionable accuracy, meaning that Boltz-2's binding-affinity module doesn't work well for large beyond-rule-of-5 molecules like peptides, bifunctional degraders, or oligonucleotides.
5.8 Performance and training concerns
Several scientists have voiced concerns about the physicality of Boltz-2's predictions. In a post on X, John Parkhill (Terray) wrote:
because of the vast data imbalance in the public data, inferring across uniform chemical space yields an obviously unrealistic distribution...
In response, Gabriele Corso (an MIT graduate student and one of the Boltz-2 authors) wrote that the pIC50 output "is not supposed to be used on arbitrary chemical spaces but only for hit-to-lead stage compound series" and said that the hit/decoy head should be used instead.
In the Boltz-2 launch presentation at MIT, Pat Walters voiced concerns about the training split used in the work, noting that assessing overall sequence similarity may lead to substantial pocket-level train–test data leakage (source).
Olexandr Isayev recently alleged on X that the performance on unreported targets was "much more... modest" than the published results, and Saro Passaro responded that the average Pearson correlation was the same on a larger set.
Tushar Modi and co-workers recently evaluated Boltz-2 on a variety of protein targets: KRAS G12C, SARS-CoV-2 Mpro, PIK3-α, DHX9, cGAS, and WRN helicase. Their results were mixed; while Boltz-2 can be effective for some targets, the poses are often unrealistic, and large conformational changes are poorly described. Generalization remains an open question.
On LinkedIn, Semen Yesylevskyy reports results for Boltz-2 on the PL-REX database (originally developed to score the SQM2.20 semiempirical binding-affinity-estimation method). Semen says that Boltz-2 is "only 5-7% better than the closest ML competitor" and "only an incremental improvement" rather than a revolution.
6. What data was Boltz-2 trained on?
Boltz-2 was built on the same core structural corpus as Boltz-1—filtered RCSB/PDB entries plus ~1M OpenFold self-distillation targets with paired MSAs—but adds every protein-, RNA- and DNA-ligand complex deposited through early 2025 and systematically augments them with small-molecule symmetry and template metadata.
To teach its new affinity head, the team aligned ≈ 3M standardized Ki/Kd/IC50 measurements from ChEMBL, BindingDB, PDBbind and MF-PCBA to the corresponding targets, yielding ~750K high-quality protein–ligand pairs after noise and overlap filtering.
7. Given that public binding-affinity data is low-quality, how reliable are the predictions?
The team applies extensive curation (discussed in the paper), yet they note that there are "multiple sources of systematic noise and artifacts" in high-throughput-screening data, not all of which can be removed. Roughly 40% of "hits" predicted by Boltz-2 are false positives—so there's still plenty of errors in the Boltz-2 predictions, despite the advances presented in the paper.
In practice, this means that further validation and screening will be needed to support Boltz-2 predictions. While Boltz-2 is a big advance, it is not a substitute for a full computer-aided-drug-design workflow.
8. Does Boltz-2 eliminate all steric clashes and stereochemical errors?
Boltz-2x inherits the previously reported Boltz-1x steering potentials that can be enabled to reduce clashes. Still, the paper reminds readers that deep-learning co-folding methods can produce "incorrect bond lengths and angles, incorrect stereochemistry at chiral centers and stereobonds and aromatic rings predicted to be non-planar"—as always, use caution and visually check results for sanity and adherence to chemical principles.
9. Has Boltz-2 made free-energy (FEP/ABFE) calculations obsolete?
No! Boltz-2 approaches FEP accuracy on several public datasets while being >1000× faster (key word "approaches"), but it does not consistently match the chemical accuracy achieved by well-tuned FEP protocols, especially for subtle relative potency shifts in lead optimization. (Read more about free-energy perturbation here.)
In addition, it's not yet clear how Boltz-2 will fare on targets far outside the training data; the performance on internal targets from Recursion was worse than other benchmarks, suggesting there may be cryptic data leakage or other confounding factors. (John Taylor has raised these concerns on LinkedIn.)
Early external benchmarks suggest that Boltz-2 can rank compounds well but sometimes struggles at quantitative affinity prediction; as more benchmarks are run and published, we expect that the field will gain a much clearer understanding of Boltz's strengths and weaknesses.
10. What hardware do I need to run Boltz-2?
Boltz-2 can allegedly be run on CPU, GPU, or TPU hardware. By default, Boltz-2 expects a GPU to be present, but this can be changed through the --accelerator setting. Inference on CPU-only machines is reported to be very slow.
In the provided paper, the authors run inference through H100 GPUs; older GPUs like V100s may require disabling trifast kernels (see this GitHub issue). Performance will likely be worse on older GPUs, and in some cases GPUs may have insufficient memory.
At the time of writing there is no support for Apple Silicon MPS hardware acceleration, although an open pull request may solve this problem.
10. How is Boltz-2 licensed, and how can I cite the model?
The Boltz-2 code, weights and data are released under the MIT license, an open-source license with permissive redistribution rights.
If you use Boltz-2, please cite:
Passaro S. et al. "Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction" (2025).
The Boltz-2.1 model is a closed-source model distributed by Boltz PBC (the startup).










