Tracking External Boltz-2 Benchmarks
by Corin Wagen · July 1, 2025
Three weeks ago, a team of scientists from MIT and Recursion released Boltz-2, a co-folding model which not only predicts the structure of bound protein–ligand complexes but also "approaches the accuracy of FEP-based methods" for binding-affinity prediction. This is an extraordinary claim, and one which prompted thousands of scientists (including us) to start investigating Boltz-2 for structure-based drug design. (For a more detailed look at how Boltz-2 works and the potential uses, read our full FAQ.)
(Free-energy perturbation, often abbreviated FEP, is the "gold standard" method for predicting protein–ligand binding affinity.)
Over the past few weeks, a variety of scientific teams have disclosed external benchmarks of Boltz-2. This field is moving incredibly fast, so these benchmarks are hard to keep track of: some happen on LinkedIn, while others are on X or various blogs around the Internet. To make it easier for our users to keep track of the latest updates surrounding Boltz-2, we've compiled the most relevant data on this page. Although it's still early—it hasn't even been a month since Boltz-2 was released—the model's strengths and limitations are gradually becoming clear. (Note: we're excluding random posts of single structures here, since most of these lack clear systematic comparisons to experiment.)
This is a living document, and will be updated as additional benchmarks are released. This page last updated March 9, 2026.
These benchmarks predate the June 2026 release of the new Boltz-2.1 model, which contains changes to the binding-affinity-prediction head.
PL-REX Benchmark (Semen Yesylevskyy)
This benchmark, posted on LinkedIn a week ago, evaluates the performance of Boltz-2 against a variety of physics- and ML-based methods on the 2024 PL-REX dataset. This is a "best case" scenario for physics-based methods, since the protein–ligand complex is known with relatively high confidence for these systems.
Yesylevskyy compared the Pearson correlation coefficient of all methods for ranking the relative affinity of different binders. He found that the SQM 2.20 method (for which the PL-REX dataset was developed) significantly outperformed all other methods, with Boltz-2 coming in second place.
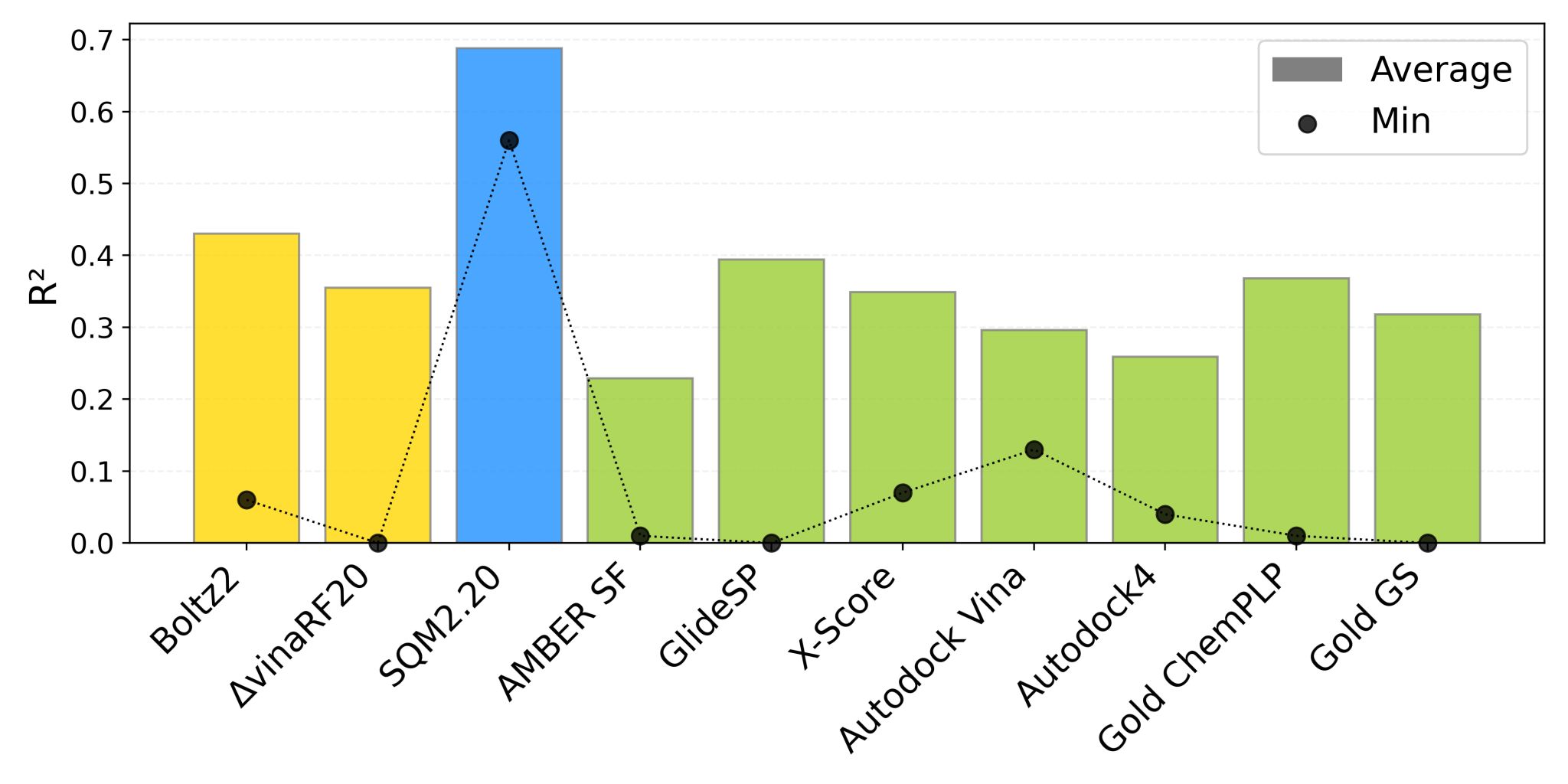
Comparison of a variety of methods on the PL-REX binding-affinity benchmark.
Here's what Yesylevskyy has to say about this:
Boltz-2 scores the second being only 5-7% better than the closest ML competitor ΔvinaRF20 and the closest physics-based competitors GlideSP and Gold ChemPLP. Boltz-2 is still far cry below SQM2.20 and only reaches mean correlation of ~0.42 with experimental values... So, according to this test, Boltz-2 is only an incremental improvement over existing affinity prediction techniques rather than a revolution. Moreover, its inference speed was rather disappointing in our tests being an order of magnitude slower than conventional docking programs such as Vina or Glide.
It's worth noting that although SQM 2.20 performs well on this benchmark, a similar semiempirical method was recently shown to perform poorly on the ULVSH virtual screening dataset.
Uni-FEP Benchmark (Xi Chen)
On LinkedIn, Xi Chen and co-workers from Atombeat recently disclosed benchmark results for Boltz-2 on the Uni-FEP dataset. This benchmark set comprises approximately 350 proteins and 5800 ligands.
Chen reports that Boltz-2 gives "consistently strong results — measured by both correlation terms and mean error terms— across 15 protein families," including cases where conformational effects are significant, like GPCRs and kinases. Unfortunately, Boltz-2 significantly lagged FEP in cases where buried water was known to be important, a sign that these effects are not implicitly accounted for by the model:
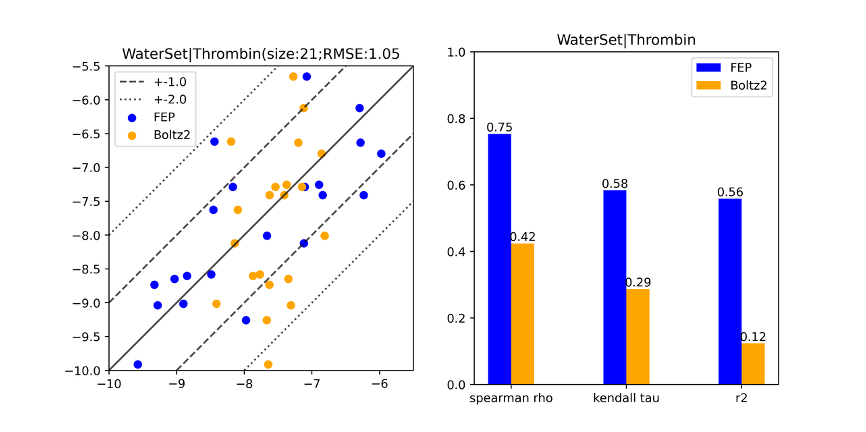
Comparison of Boltz-2 to FEP in cases where buried water is important.
Another interesting observation is that Boltz-2 consistently underestimates the spread of binding affinities present in experimental data. In the below two cases, the predicted range of binding affinities is significantly tighter than either the observed experimental values or the predictions from the conventional physics-based FEP workflow:
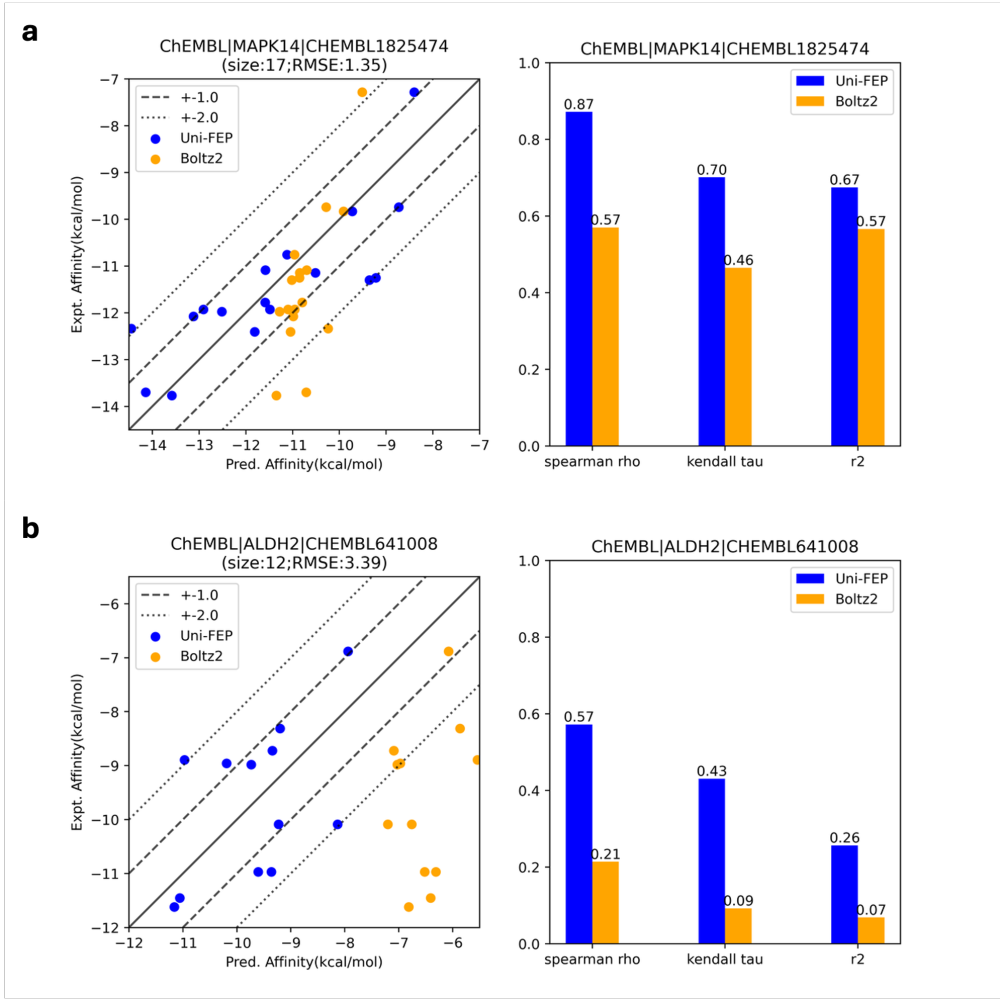
Comparison of Boltz-2 to FEP, illustrating the propensity of Boltz-2 to compress affinity values.
Here's what Chen has to say:
One general trend we observed — independent of specific targets — is Boltz-2's tendency to predict binding affinities within a narrow range, typically within 2 kcal/mol. Figures 5a and 5b illustrate examples. We found this behavior on 75 of the 350 targets evaluated. For 21 of those, the experimental binding affinities spanned more than 4 kcal/mol — yet Boltz-2 clustered predictions near the mean, effectively "regressing to the center."
Similar observations were recently reported by John Parkhill on X.
Six Protein–Ligand Systems (Tushar Modi)
Tushar Modi and co-workers at Deep Mirror recently disclosed benchmarks for six protein–ligand systems. Their overall conclusions were that Boltz-2 did well for stable and rigid systems, but struggled with ligand geometries or in cases where conformational flexibility was important:
Boltz-2 often has difficulty when a protein must undergo a big shape change or has multiple mobile domains with little precedent in the training data. If a protein needs to bend into a new shape to accommodate a ligand (like the allosteric changes in PI3K-α or WRN, or the dynamic binding required in cGAS), the unguided model usually fails to predict that rearrangement. These cases often require additional help—such as supplying a template of the alternate conformation or running a refinement step—to obtain the correct pose.
Note that this conclusion is the exact opposite of what Xi Chen noted above.
ASAP-Polaris-OpenADMET Challenge (Auro Varat Patnaik)
Auro Varat Patnaik, a graduate student at the University of Edinburgh, ran a retrospective analysis of how Boltz-2 would have performed on the ASAP-Polaris-OpenADMET antiviral challenge. He found that Boltz-2 performed very poorly, with a mean absolute error worst among any method studied.
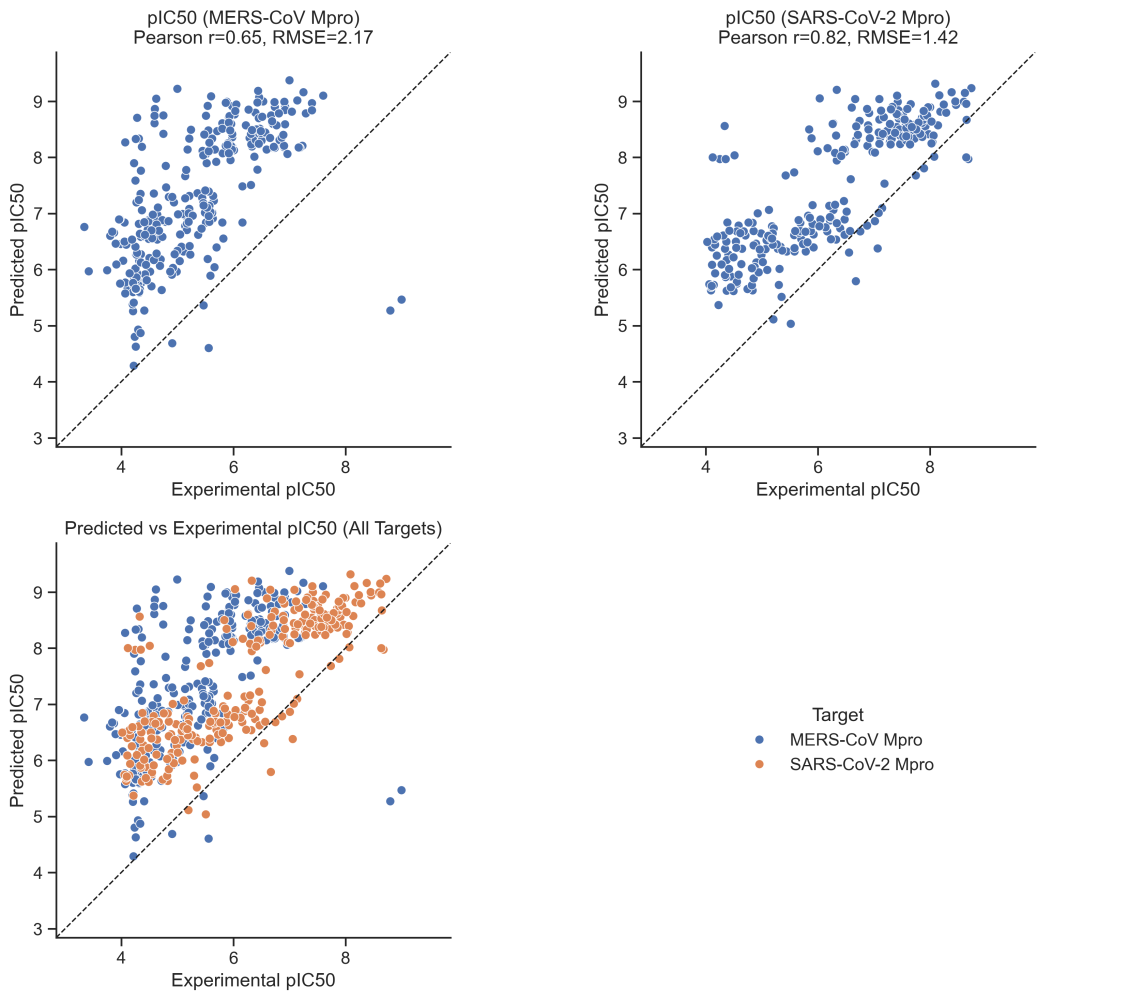
Comparison of Boltz-2 predicted pIC50 values to experimental values on the ASAP-Polaris-OpenADMET challenge.
Patnaik offers the following caveat:
Compared to the other methods, a vanilla BOLTZ-2 seems to be far behind, but it's critical to note that the competing methods were fine-tuned models. A fine-tuned BOLTZ-2 could potentially provide much better results.
At a minimum, it seems that zero-shot Boltz-2 is not a replacement for fine-tuned methods using target-specific data.
Molecular Glue Binding Affinity (Dominykas Lukauskis)
Dominykas Lukauskis and co-workers from Ternary Therapeutics compared the performance of Boltz-2 and FEP (using OpenFE) on a set of 93 molecular glues with experimentally determined ternary-complex binding-affinity data. They found that Boltz-2 dramatically underperformed FEP, showing "generally poor or even negative correlations" and large absolute errors, despite generally good structural validity and accuracy of the predicted complexes.
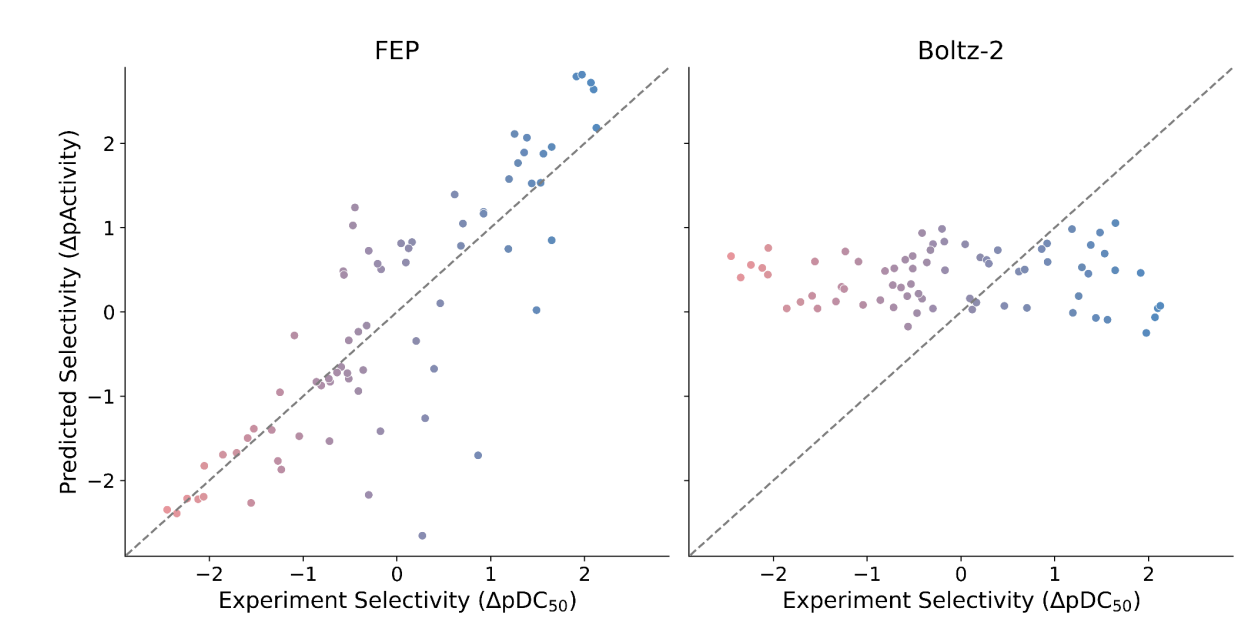
Comparison of Boltz-2 predicted affinities to experimental values on the Ternary Therapeutics dataset.
In their own words:
The poor performance of Boltz-2 suggests it is not suitable for high-throughput screening of molecular glues, highlighting the need for more accurate, high-throughput machine learning methods for pre-FEP screening
ULVSH Classification (Didier Rognan)
Rognan and co-workers recently studied the performance of Boltz-2 on their previously reported ULVSH screening dataset and found that Boltz-2 outperformed other studied methods at differentiating binders from non-binders. However, the authors also found that Boltz-2 was able to identify binders even after adversarial mutations designed to eliminate key binding-site interactions, suggesting that Boltz-2 might be getting "the right answers for the wrong reasons" without actually learning the underlying physics of protein–ligand binding. (These results mirror previous work from Matthew Masters and co-workers showing that co-folding methods do not learn correct physics.)
Memorization Benchmark (Mrinal Shekhar)
Recent work by Shekhar and co-workers studied whether the "impressive enrichment performance" of Boltz-2 could be attributed to "genuine understanding of protein–ligand interactions" or simple memorization of known systems and motifs. To assess this, the authors conducted virtual knockout experiments where they purposefully ablated the binding site of known complexes via alanine scanning, steric hinderance (replacing all residues with phenylalanine) or polarity and charge inversion (replacing positively and negatively charged residues). While these mutations resulted in negligible enrichment for physics-based methods like docking, Boltz-2 retained high enrichment, demonstrating "profound and systematic memorization artifacts in Boltz-2" and calling into question the utility of Boltz-2 in prospective virtual-screening scenarios.
Influence of Ligand Charge (Alisa Bugrova)
Alisa Bugrova and co-workers studied how changing the charge of small molecule ligands (specifically methylammonia and acetic acid) affected their geometry, as predicted by Boltz-2 and other co-folding methods (e.g. Chai-1 and AlphaFold 3). Surprisingly, the authors found that inputting these ligands as SMILES vs CCD led to significant differences in their predicted structure: "Boltz-2 predictions are extremely sensitive to the input format." This suggests that the models are not fully learning the underlying physics, since in theory SMILES vs CCD representations of the same ligands should lead to identical results.
Large-Scale Reliability Testing (Shunzhou Wan)
Shunzhou Wan, Peter Coveney, and co-workers investigated the performance of Boltz-2 using large-scale TNKS2 and 3CLPro datasets, comparing predicted geometries to experimental & docked poses and predicted binding affinity values to MD-based estimates following the ESMACS protocol. The authors found that the accuracy of the predicted geometry was highly variable, with many ligands having >10 Å RMSD error in the predicted pose. Additionally, the geometry of the predicted poses was often of dubious quality, with "significant physical issues" like ring de-planarization and oversaturation of aliphatic chains.
Wan and co-workers also found that the predicted binding-affinity values had only "weak to moderate correlations with physics-based free energy values," rendering them useless in a case study of the top 100 compounds. The authors note that the significant run-to-run variability of Boltz-2 (up to 1.5 kcal/mol) may drown out any potential signal that could be used for enrichment, concluding that Boltz-2 "cannot serve as a reliable substitute for high precision, physics-based methods in late-stage drug discovery."
Conclusions
While this field is moving fast, some tentative conclusions can be drawn. Here's our current thinking on Boltz-2:
- Boltz-2 can be reproducibly better than conventional protein–ligand docking for in-distribution tasks.
- However, it struggles in complex cases or cases that are poorly represented in the training data. It's still not 100% clear what these cases are; some benchmarks allege that flexible systems perform badly, for instance, while others disagree.
- Boltz-2 is not yet a replacement for "gold-standard" physics-based methods like FEP or fine-tuned target-specific methods.
When used properly, it's likely that Boltz-2 can be a very useful tool in the drug-discovery arsenal; but it's not a solution in isolation, and likely needs to be embedded in a proper virtual-screening workflow to give useful results.
Addendum: Chai-2
Yesterday, Chai-2 was released. Although minimal technical details were disclosed, Chai-2 appears to be a co-folding-based workflow involving a sequence of models and physics-based steps that can be used for zero-shot antibody design. In combination with Adaptyv Bio, the Chai-2 authors reported a 50% wet-lab success rate against a panel of 52 diverse protein targets; the full technical report gives more target details.
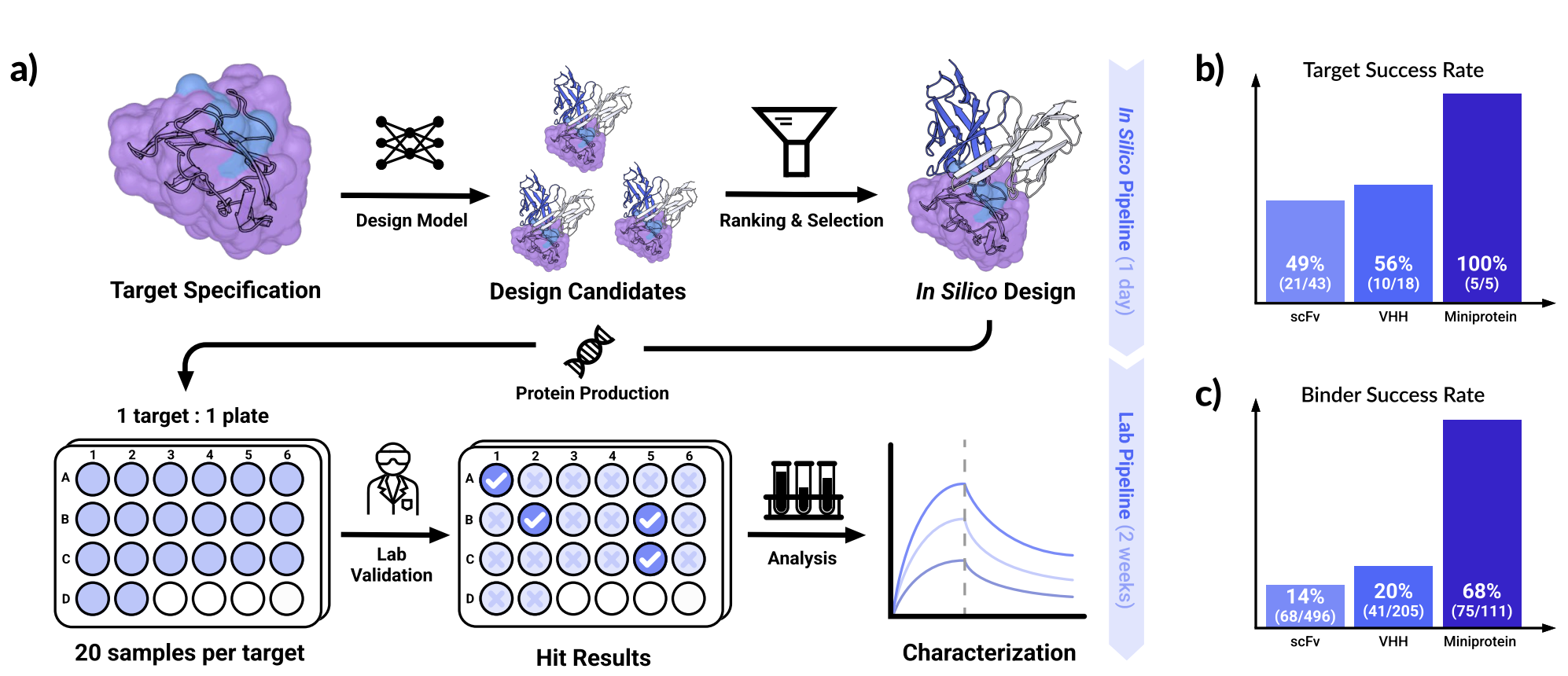
Figure 1 from the Chai-2 technical report.
Since Boltz-1 and Chai-1 were virtually clones, it's interesting to reflect on the ways these two projects have evolved. Boltz-2 has focused on small molecules and binding-affinity prediction within a single model, while Chai-2 has expanded into an entire end-to-end pipeline and seems to be focusing on antibody/nanobody design. It will be interesting to see where both projects go next!










