How to Design Protein Binders with BoltzGen
by Corin Wagen and Ari Wagen · Oct 27, 2025
BoltzGen is a comprehensive protein binder design pipeline built on top of the Boltz model family. It uses BoltzGen (a diffusion model for structure generation), BoltzIF (for inverse folding), and Boltz-2 (for structure validation and affinity prediction) to generate folded designed protein structures (whether proteins, peptides, or nanobodies) to bind given small molecule, protein, or nucleotide targets. BoltzGen was developed by Hannes Stärk and co-workers from MIT and other institutions; for full details, see the paper or the repository.
At a high-level, you can use BoltzGen by defining one or many targets that you want to design binders for, inputting any constraints you want enforced during the design process (like sequence length), and then running the BoltzGen pipeline.
How BoltzGen Works
BoltzGen supports four different default protocols:
protein-anythingfor designing proteins to bind protein/peptide targetspeptide-anythingfor designing peptides to bind protein targetsprotein-small_moleculefor designing proteins to bind small moleculesnanobody-anythingfor designing single-domain antibodies or "nanobodies"
These protocols all operate similarly—BoltzGen designs binders by proceeding through seven discrete steps:
- Design. Design initial binder structures according to input specification using a diffusion model (using the core BoltzGen model).
- Inverse folding. Predict sequences of amino acids that will fold into those structures ("inverse folding" using the BoltzIF model).
- Design folding. Refold the newly-predicted amino-acid sequences with their targets using Boltz-2 to validate that they're actually predicted to bind the target.
- Folding. Refold standalone structures of newly-predicted amino-acid sequences (skipped if using the
peptide-anythingornanobody-anythingprotocols) to validate that the designed proteins will be stable on their own. - Affinity. Predict protein–ligand binding affinities (if using the
protein-small_moleculeprotocol). - Analysis. Analyze to predict design quality.
- Filtering. Filter and rank designs to select best candidate binders.
The BoltzGen workflow is intentionally very tunable. In an interview, first author Hannes Stärk urged scientists to experiment with BoltzGen's settings:
Treat BoltzGen as an iterative design partner. Start small, inspect your results, and adjust parameters. Don't just go with the pre-sets. Explore different binding sites or constraints, rerun your designs, and compare. We've intentionally exposed many control options — binding-site flexibility, sequence length, exclusion zones — so users can see how changes affect outcomes.
Input Design Specification
When creating a YAML design specification for running the BoltzGen pipeline, there are two top-level keys: entities and constraints. There are three types of "entity": protein, ligand, and file. Constraints define limits applied to the system. BoltzGen supports two constraint types: covalent bonds (bond), which defines a bond between two components, and total sequence length (total_len), which sets an upper bound on the combined sequence length of the complex.
Here's a few more details about the specific types of entities supported in BoltzGen and how to specify complex input structures. An exhaustive list of all inputs can be found in the BoltzGen repository.
Protein
id: a unique identifier for the protein chain, often a letter.sequence: the amino acid sequence; use number ranges (e.g.140..180) to specify that BoltzGen should generate a binder of that length, and use specific sequences (e.g.,MKTAYIAKQ) when the sequence is fixed.secondary_structure: an optional input which specifies the desired secondary structure of the protein.binding_types: a string or dictionary that defines which residues should be involved in a binding interaction.cyclic:Trueif the protein/peptide is cyclic andFalseotherwise.
Ligand
id: a unique identified for the ligand.ccd: the Chemical Component Dictionary identifier, only present for common ligands.smiles: the SMILES associated with the ligand.binding_types: as before, specifies if the ligand should be involved in a binding interaction.
File
path: the path to the file.include/exclude: which components of the file to import.msa: the path to the multiple-sequence-alignment (MSA) data.binding_types: as before, defines which residues should be involved in a binding interaction.secondary_structure: as before, specifies the desired secondary structure of the protein.
Command-Line Options
In addition to the above structure-input method, BoltzGen also supports a variety of command-line options. The full list is documented on Github; here's a short list of some of the most useful.
--output: where the output files should go.--devices: which devices (i.e. GPUs) will be used.--num_designs: how many designs will be generated initially.--budget: the number of diverse, high-quality designs you'll ultimately receive from the full set of--num_designsintermediate designs, typically 10-100.--alpha: controls the tradeoff between diversity and quality for final design selection: settingalphato 0.0 ignores diversity and chooses only the highest-quality designs, while settingalphato 1.0 chooses designs solely based on diversity.alphadefaults to 0.01 for peptides and 0.001 for all other protocols.--steps: which of the seven steps to run (e.g.inverse_foldingto only run the inverse-folding step).--additional_filters: allows for additional filters to be applied at runtime.
Outputs
After following any of the above protocols, BoltzGen outputs a ranked, filtered set of designs in a user-specified directory. The outputs can optionally be re-filtered and re-ranked by running boltzgen run --steps filter, which the authors recommend.
Examples
Here's some representative examples from the BoltzGen team with the corresponding design specification YAML file. Additional input files can be found by following the provided links to the BoltzGen GitHub page.
Designing a Protein to Bind Brilacidin
This input file specifies brilacidin, a small-molecule antibiotic, via SMILES and requests a 140–180 residue protein that will bind it. For this input file, the protein-small_molecule protocol should be employed.
entities:
- protein:
id: A
sequence: 140..180
- ligand:
id: B
smiles: "C1CNC[C@@H]1OC2=C(C=C(C=C2NC(=O)C3=CC(=NC=N3)C(=O)NC4=CC(=CC(=C4O[C@@H]5CCNC5)NC(=O)CCCCN=C(N)N)C(F)(F)F)C(F)(F)F)NC(=O)CCCCN=C(N)N"
Designing a De Novo Zinc Finger to Bind DNA
This input file loads DNA in from the provided CIF file and requests a 40–120 residue protein to bind it. For this input file, the protein-anything protocol should be used.
entities:
- protein:
id: G
sequence: 40..120
- file:
path: zf.cif
include:
- chain:
id: C1
- chain:
id: B1
Designing a Cyclic Peptide to Bind KRAS
This input loads the KRAS structure from the 8JJS PDB entry, loading only chains A and C to remove the existing binder. Specific residues are specified to denote the intended binding site, and an 8–16 amino-acid cyclic peptide is requested. For this input file, the peptide-anything protocol should be used.
entities:
- protein:
id: B
sequence: 8..16
cyclic: true
- file:
path: 8jjs.cif
include:
- chain:
id: A
- chain:
id: C
binding_types:
- chain:
id: A
binding: 12,14,61,63,73,76,77,83,101,104,108
Designing a Peptide to Bind to Disordered Protein Regions
Here, a protein is loaded from a CIF file. The disordered end of the protein is loaded as a group with visibility 0, meaning the structure is not specified, and these disordered residues are labeled as the binding region. A oligopeptide with 12–20 amino acids is requested as a binder; for this input file, the peptide-anything protocol should be used.
entities:
- protein:
id: B
sequence: 12..20
- file:
path: cryptochrome4_european_robin_bird_boltz_prediction.cif
include:
- chain:
id: A
structure_groups:
- group:
visibility: 1
id: A
- group:
visibility: 0
id: A
res_index: 494..
binding_types:
- chain:
id: A
binding: 494..507
Miscellaneous Notes
Cysteines
Cysteine is a very reactive amino acid, and cysteine residues are relatively rare in naturally occurring proteins (and often serve a specific purpose). For this reason, BoltzGen does not include Cys residues in the peptide-anything or nanobody-anythingprotocols by default, although this can be overridden. (This is common; BindCraft also excludes Cys residues, for instance.)
Ubiquitin
Asking BoltzGen to design a protein in the 73–76 amino-acid range frequently results in BoltzGen returning ubiquitin or very similar structures. The authors attribute this to the abundance of ubiquitin in the PDB, and plan to fix this in the future—for now, just double-check that whatever you've designed isn't ubiquitin if you asked for a binder with 70–80 amino acids.
External Validation
As of October 2025, BoltzGen is a recently released tool. While the BoltzGen paper includes expensive experimental validation carried out in collaboration and industry, it's worth noting that BoltzGen does not yet have published external validation data from third parties, unlike older tools like BindCraft or RFdiffusion. Users should be aware they are adopting cutting-edge but less battle-tested technology.
Speed
The BoltzGen GitHub recommends generating between 5,000 and 60,000 designs for production use. How long does this take? The exact speed obviously depends on the system size; in our hands, BoltzGen takes roughly 30–60 seconds per design for systems with a few hundred amino acids but can be substantially slower for large proteins. It's difficult to generate more than 100 designs per GPU-hour except for very small systems. (These timings are for modern GPU-enabled systems; with older GPUs, calculations may run substantially slower.)
Accordingly, running a single production-scale binder-design campaign is a non-trivial investment of computational resources. It's worth noting, however, that running an experimental binder-design campaign would be even more expensive!
Running Locally
1. Install boltzgen Locally
To run BoltzGen locally, first install the boltzgen package following the instructions on GitHub.
2. Generate the YAML Design Specification
Following the format detailed above, generate a YAML design specification to tell BoltzGen what to design.
3. Run boltzgen
Use the boltzgen run command with any requested options. This command submits a YAML design specification at design_specification.yaml, writing outputs to example_output/ and generating 10 designs following the protein-anything protocol towards a final budget of two designs. (As discussed above, this is a very low number of designs, and should be viewed as a test run rather than a full-fledged production protein-design calculation.)
boltzgen run design_specification.yaml \
--output example_output \
--protocol protein-anything \
--num_designs 10 \
--budget 2
(This step may take a while to run.)
4. Analyze the Results
Visualize the provided designs using a web-based or local protein viewer. You can optionally re-filter the designs using the boltzgen run --steps filtering command.
Running Through Rowan
1. Select Rowan's "Protein Binder Design" Workflow
Log into Rowan, navigate to an existing project, and select "Protein Binder Design" from Rowan's workflows.
2. Input Structures and Design Specification
All three entity types can be added through Rowan's interface. Rowan's existing protein-editor functionality makes it easy to sanitize PDB files and remove existing chains, binders, or small-molecule ligands. The below specification requests 8–16 amino-acid cyclic-peptide binders to an uploaded KRAS G12D structure.
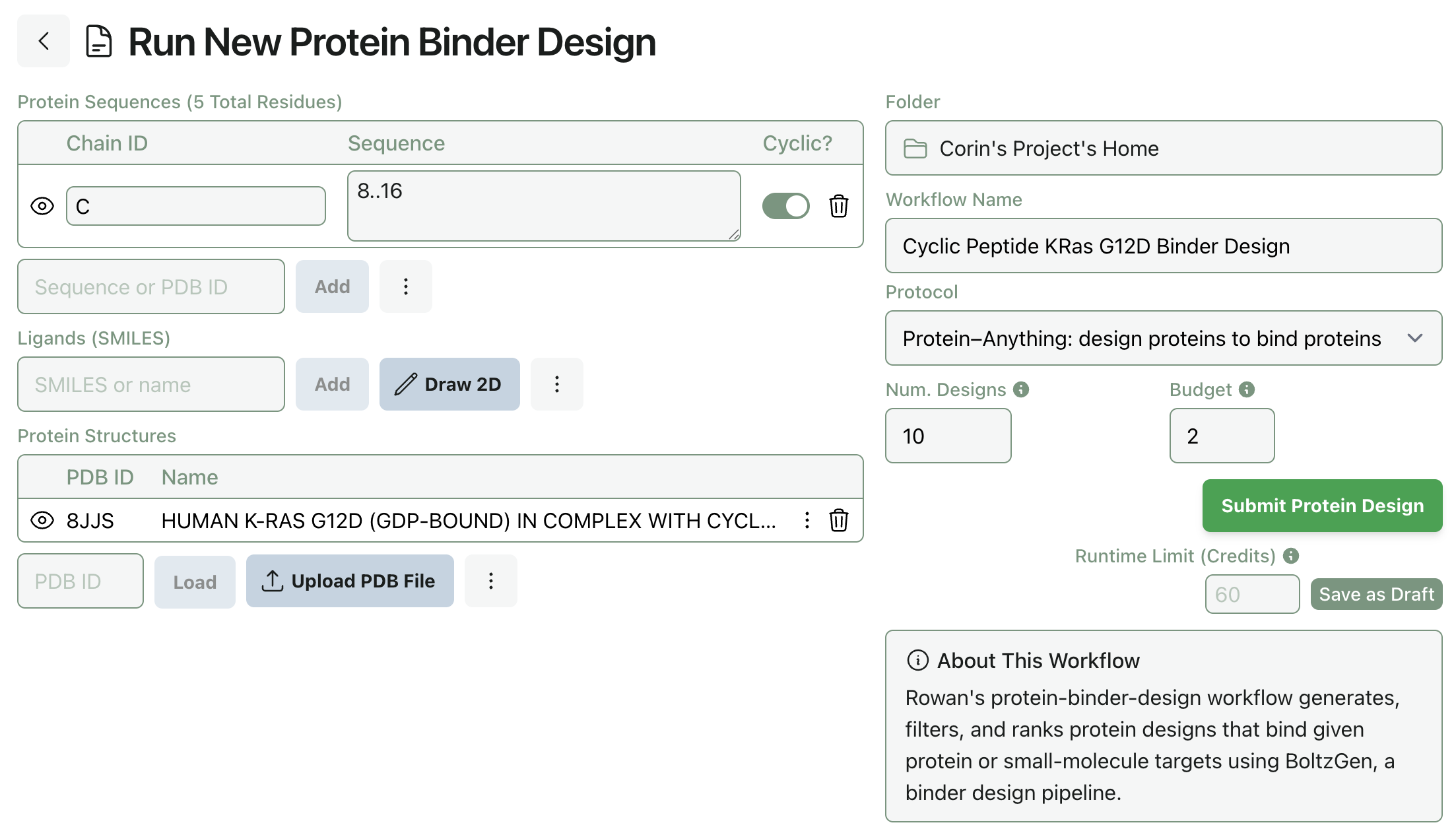
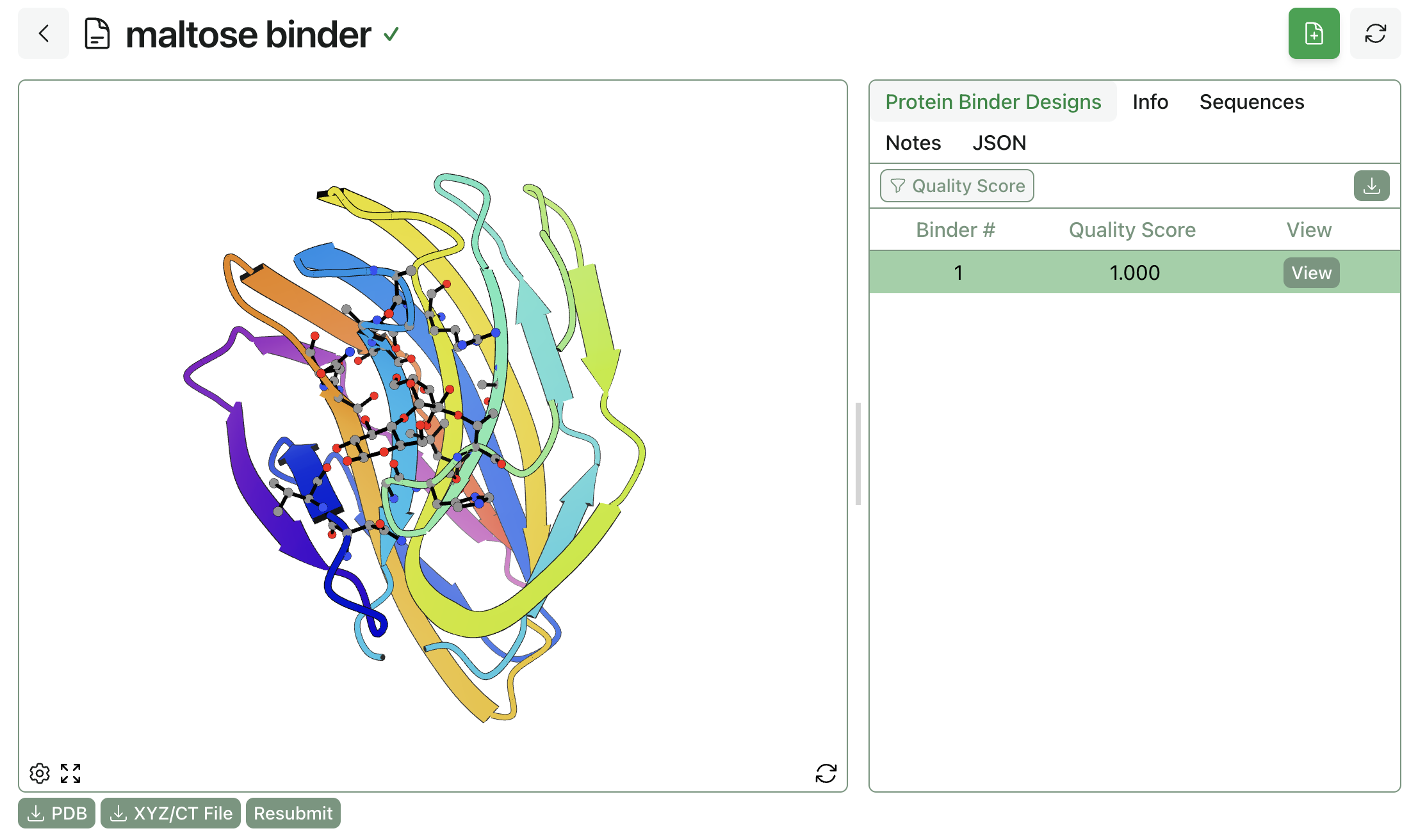
3. Submit and View Results
Rowan automatically allocates computational resources, runs the requested BoltzGen workflow, and saves results to the database. The generated binders can be viewed through Rowan's web interface. Here's an example of a maltose-binding protein:
Why Use BoltzGen?
BoltzGen is an efficient all-in-one solution for a diverse array of protein-binder-design problems: de novo small-molecule binder design, protein–protein binder design, and design of oligopeptide / cyclic-peptide binders, to name just a few of the many uses. BoltzGen is modular and easily configurable, making it easy for scientists to quickly iterate on various designs, and also integrates the state-of-the-art Boltz-2 protein co-folding model. For small-molecule use cases, BoltzGen also allows for binding affinities predicted by Boltz-2 to be used in binder prioritization.
By offering an all-in-one pipeline with predefined protocols for each modality, BoltzGen makes it easy to run state-of-the-art protein-design workflows without needing to build specialized infrastructure or become a domain expert. BoltzGen can be run directly through Rowan's GUI, is automatically integrated with our protein visualization and analysis tools, and can be tried for free.
Further Reading
Protein binder design is a very active area of computational biology! If you're interested in reading more about computational binder design, we enjoyed reading "Minibinder design isn't that hard" by Nick Boyd and Sam Guns at Escalante Bio.
If you're interested in testing different models in this space, we recommend checking out:
- BindCraft, a state-of-the-art binder design pipeline from Paseca and co-workers at EPFL and other institutions. (There's also FreeBindCraft if you don't have a Rosetta license.)
- LigandMPNN, a protein sequence design model from Dauparas and co-workers at the Baker lab.
- Germinal, an antibody-design package from Mille-Fragoso and co-workers at Stanford.
- Mosaic, a modular protein-design package from the team at Escalante Bio.
- BAGEL, a protein-engineering package from Jakub Lála and co-workers at Imperial College.
Chai Discovery also reported successful de novo protein binder design results in their technical report about the closed-source Chai-2 model.










