Case Studies with Rowan's Hydration-Site Analysis
by Ishaan Ganti and Corin Wagen · July 16, 2026
We've recently added automatic hydration-site detection capabilities to Rowan's pose-analysis molecular-dynamics workflow, which makes it easy to find conserved waters in protein–ligand complexes and distill less-obvious insights out of MD trajectories. To verify that our hydration-site-detection methodology is performant and accurate, we wanted to benchmark our detection on systems with thoroughly studied water dynamics.
Benchmarking hydration-site detection is tricky, since there isn't a single obvious "ground truth" metric to evaluate against (unlike, for instance, FEP). There are many different implementations of hydration-site detection, like Schrödinger's WaterMap and OpenEye's SZMAP, each of which makes different methodological choices and thus give slightly different answers for valid reasons.
To benchmark our hydration-site detection implementation, we re-examined three notable protein–ligand case studies to confirm that Rowan's hydration-site analysis correctly finds the reported waters. We also looked at two molecular-glue systems, to demonstrate extension of this workflow to larger structures (and because water binding in these high-surface-area inferfaces is complicated and interesting), for a total of five benchmark systems.
How Rowan's Hydration-Site Detection Works
We start by heavily filtering the waters we consider to just near the interface: water oxygens within 5 Å of both a ligand atom and a protein heavy atom, per frame. Remaining waters are then considered over a 0.5 Å cubic grid superposed onto the protein–ligand complex. Each frame contributes at most one count per voxel. Dividing all counts by the total number of frames gives, for each voxel, the fraction of frames in which the voxel was occupied.
Then, we look for local-occupation maxima (peaks) above a set threshold—the threshold is to ensure that a site isn't selected from a cluster of adjacent sites that are all barely occupied. Surviving peaks are clustered greedily from the highest occupancy downwards; each unclaimed peak seeds a site and absorbs neighbors within 1.4 Å. These are the final hydration sites, though occupancies are recounted after clustering.
Sites in hand, we analyze hydrogen bonding between the closest water to each site per frame and nearby protein & ligand atoms using heuristic geometry and element checks. Waters that hydrogen-bond to the ligand and the protein simultaneously ("bridging" waters) are important interaction mediators, and as such, are explicitly listed alongside the hydration sites.
After aggregation across all replicates, each site gets a compact fingerprint: centroid, overall occupancy, bridging occupancy, and a ranked list of bridging protein residues.
1HPX: HIV-1 protease with KNI-272
Our first case study is the HIV-1 protease/KNI-272 complex, PDB 1HPX. This is a classic example where ordered interfacial waters are reported to be crucial to inhibitor binding dynamics. The original 2.0 Å crystal structure reported that KNI-272 binds through hydrogen bonds to multiple bridging waters, including one associated with the catalytic Asp125 residue. Subsequent studies confirmed that several waters at the protease–inhibitor interface are relevant to binding:
- A flap water mediating hydrogen-bond interactions between KNI-272 and the Ile50/Ile150 flap backbone amides.
- A catalytic-region water associated with Gly27 and Asp125.
- A water bridge on the Asp29-side of the inhibitor interface.
- A water bridge on the Asp129/Gly127 side of the inhibitor interface.
We ran Rowan's hydration-site analysis and successfully relocated sites 1, 2, and 4. Here's a visual overview of the results:

| Site | Occupancy % | Bridging % | Unique waters | Bridge residues |
|---|---|---|---|---|
| 1 | 97.3 | 96.6 | 1 | A:ILE50 (86%), B:ILE50 (78%) |
| 2 | 89.4 | 19.6 | 1 | B:ASP29 (20%), B:GLY27 (12%), A:ARG8 (1%) |
| 3 | 75.1 | 39.4 | 81 | B:ASP25 (39%), B:ARG8 (21%) |
| 10 | 52.2 | 33.3 | 31 | A:ASP25 (33%) |
| 12 | 40.1 | 34.2 | 3 | A:ASP25 (34%), B:ASP25 (23%) |
We find most of the reported sites, as well as a milieu of exchangeable less-occupied sites. The porous nature of this protein–protein interface means that a large number of binding-relevant waters inhabit the pocket, with varying degrees of exchange with solution; some waters seem to just be there because there's space, while other waters engage in highly conserved interactions which are almost certainly significant for binding. In particular, the single Ile50–Ile150 bridging water almost always binds to the backbone amide of KNI-272, indicating that this interaction is strongly conserved and likely important for binding.
3ATL: trypsin with benzamidine
Despite the simplicity of the trypsin–benzamidine complex, a 2022 report found that this complex has a highly structured water network. Waters in the trypsin–benzamidine complex have also been studied in other contexts previously, such as in this 2013 work on calculating binding free energies. Several key sites are documented in these two papers, including:
- A water at the bottom of the S1 pocket that forms a hydrogen-bond network enabling benzamidine release. The primary involved residues are Ser190 and Asp189.
- Water-mediated sites between benzamidine and the backbone carbonyl oxygens of Val227, Val213, and Ser214.
- A bound-pocket water connecting benzamidine to Tyr228 and Ser190 in the structured S1-pocket water network.
Rowan's hydration-site detection found the following sites:
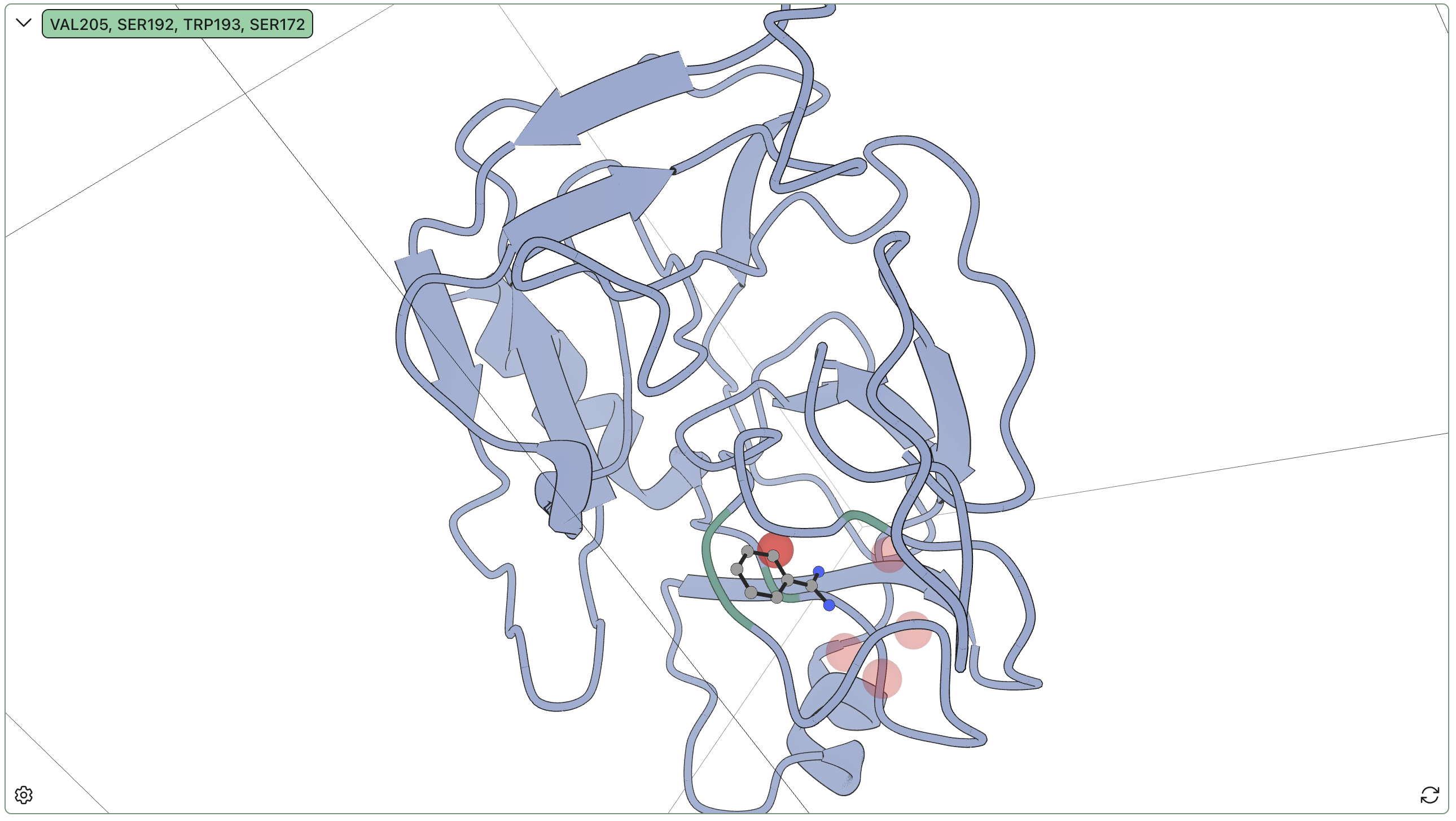
| Site | Occupancy % | Bridging % | Unique waters | Bridge residues |
|---|---|---|---|---|
| 1 | 100 | 56.2 | 1 | A:VAL205 (46%), A:SER192 (27%), A:TRP193 (20%) |
| 2 | 98.2 | 0 | 2 | — |
| 3 | 78.5 | 46.1 | 2 | A:SER195 (28%), A:LYS202 (26%), A:PRO203 (15%) |
| 4 | 78.1 | 0 | 2 | — |
| 5 | 72.5 | 0 | 6 | — |
Our site 1 is a strong match with the second site described above; the residue numbering differs between our PDB and the literature, but directly comparing the sequences allows for mapping residue labels near the binding site. Moreover, our sites 2 and 3, while more ambiguous, occupy positions very consistent with the documented important water positions in the literature. The low number of unique waters for both sites, as well as the strong hydrogen bonding in site 3, further indicate the significance of these sites.
Taken broadly, these results agree with the high-level findings from previous studies. Rather than sitting in empty space like the crystal structure suggests, the amidine group is flanked by two waters, which connect the ligand to hydrogen-bonding groups on the protein backbone. These waters are relatively anchored in place, remaining bound to the protein even when the ligand temporarily dissociates and exchanging infrequently over the simulation runtime, and are expected to be highly significant for binding.
3MXF: BRD4 inhibited by JQ1
Bromodomain-containing protein 4 (BRD4) is an epigenetic modulator that's been extensively studied in oncology contexts. In particular, there's been a lot of effort to develop small-molecule inhibitors that disrupt recognition of acetylated lysine residues on chromatin, including the famous JQ1 inhibitor developed by Jay Bradner and co-workers.
The BRD4–JQ1 complex is known to have a network of conserved water molecules in the binding pocket that are significant for inhibition. It is known that for many inhibitors in the binding pocket, a water molecule mediates a hydrogen bond between Tyr97 and the bound ligand. Our water analysis unambiguously captures this site, highlighted here in the darker red:
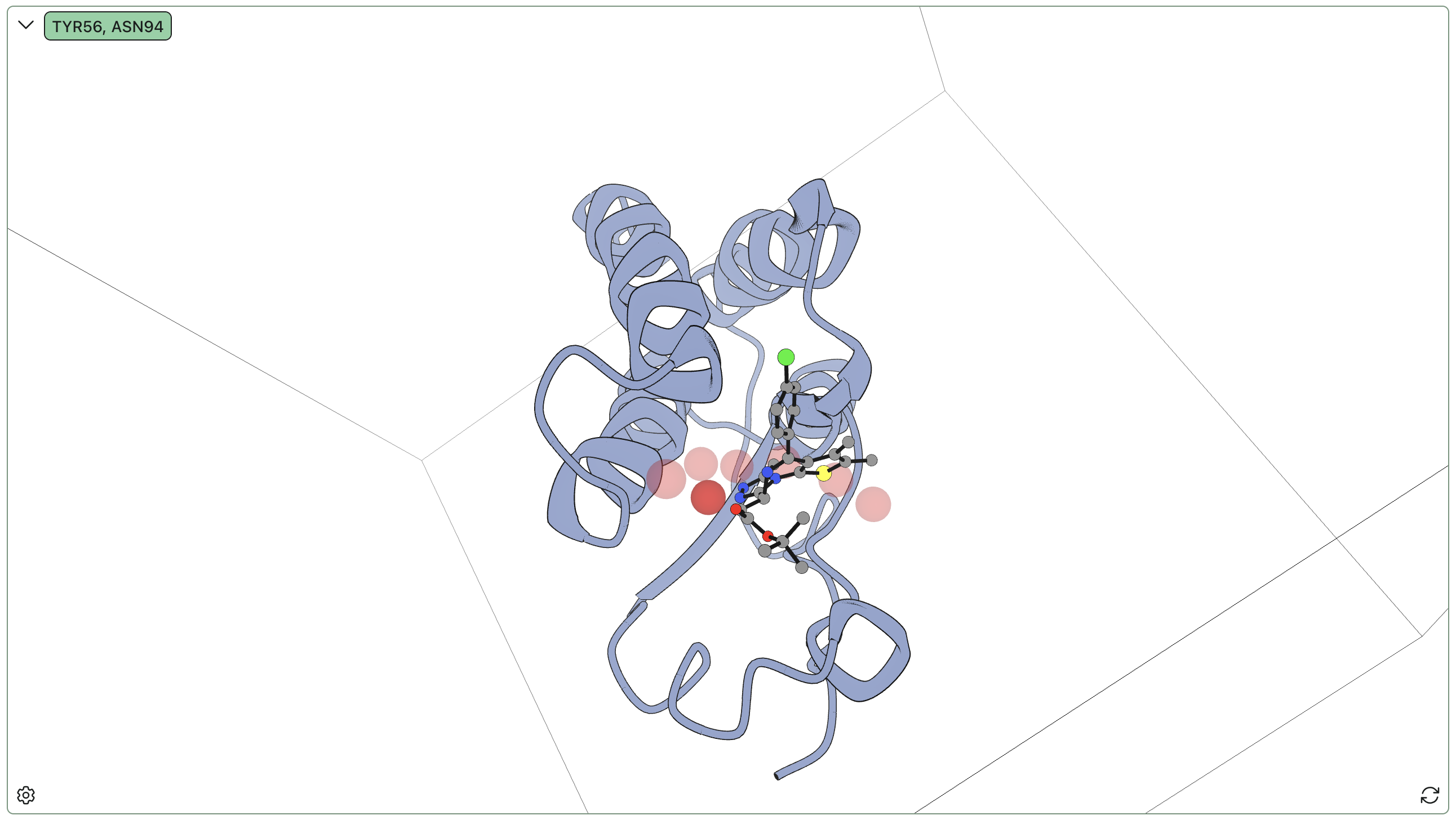
| Site | Occupancy % | Bridging % | Unique waters | Bridge residues |
|---|---|---|---|---|
| 1 | 99.0 | 0 | 1 | — |
| 2 | 94.9 | 0 | 1 | — |
| 3 | 94.2 | 0 | 2 | — |
| 4 | 93.2 | 0.778 | 1 | A:TYR56 (78%) |
Furthermore, the positions of (our) sites 1, 2, and 3 are visually consistent with known conserved waters found in crystal structures of BRD4 with various inhibitors.
The key bridging residue sits at the apex of a "water wire" comprising six conserved protein-bound waters stretching from solvent to the key Asp/Tyr residue pair. This chain of waters masks the exposed polar functionality on the back of the pocket and reshapes the pocket, explaining why e.g. docking calculations run without water struggle to reproduce the correct pose.
3FAP: FKBP12 and mTOR complex stabilized by rapamycin
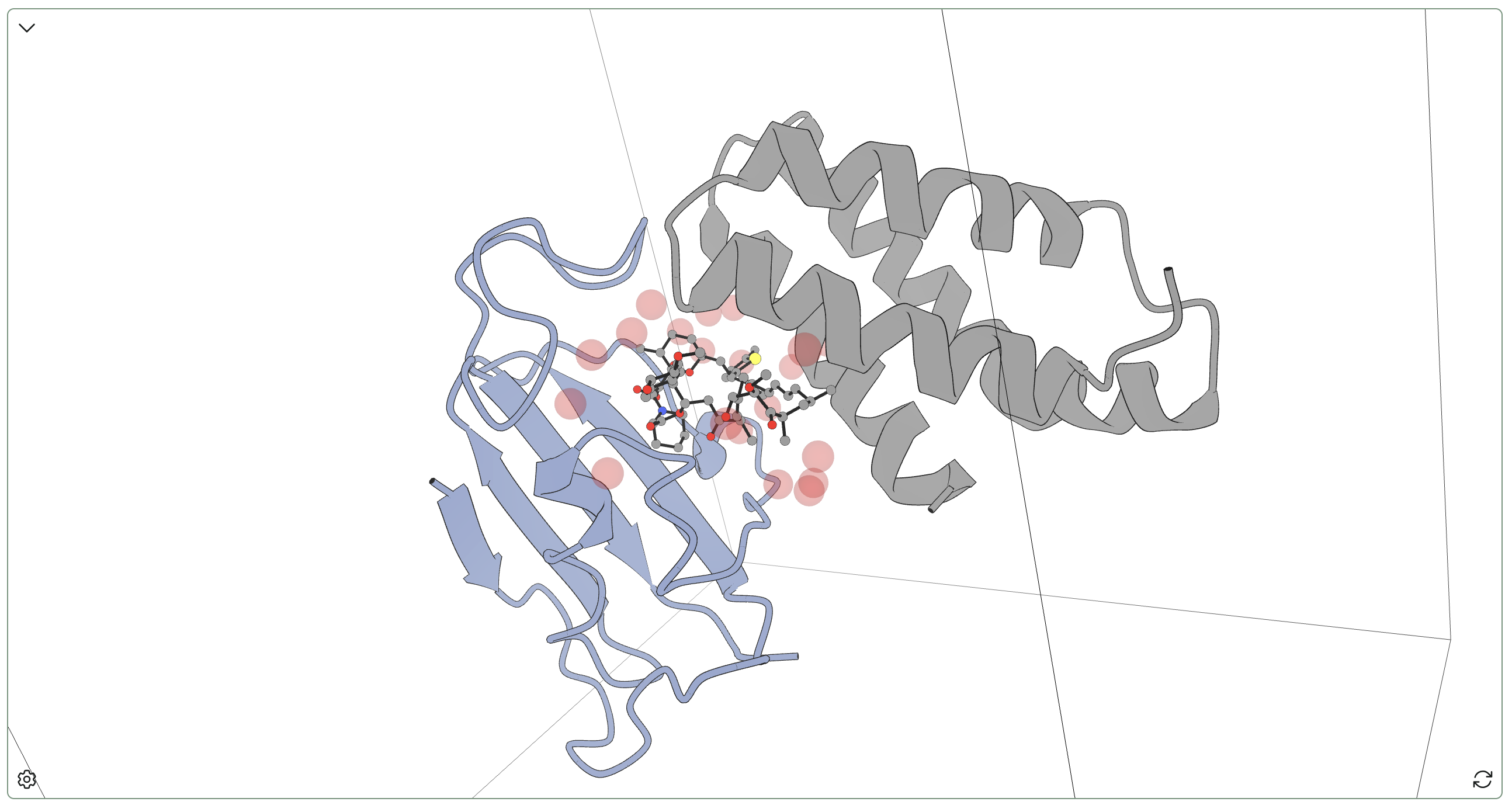
This complex is famous: rapamycin, originally discovered on Easter Island, acts as a potent immunosuppressant by stabilizing the complex of FKBP12 and mTOR (mTOR being a kinase important in various cell-cycle contexts). This interaction prevents mTOR signaling downstream of IL-2, suppressing T-cell proliferation—as such, rapamycin is widely used as an immunosuppressant that prevents organ-transplant rejection. 3FAP is the complex of FKB12, the key domain of mTOR, and a rapamycin analogue (C15-(R)-methylthienyl rapamycin).
We used Rowan's hydration-site analysis to study water dynamics in the 3FAP complex. Rapamycin (and its analogues) are large and hydrophobic compounds and accordingly there are no waters inside the macrocycle; instead, waters bind in a loose ring to the polar groups on the periphery of rapamycin. While a few waters engage in bridging interactions between rapamycin and one or both proteins (or between the two proteins), no single interaction seems particularly conserved or significant. All waters found here exchange rapidly, and few have highly conserved interactions.
This complex thus serves as a useful "negative control" for hydration-site detection: Rowan's algorithm doesn't just randomly put "conserved" waters in various places based on starting solvent structure, but can legitimately find complexes without significant waters in cases where the geometries don't lead to stable complexes.
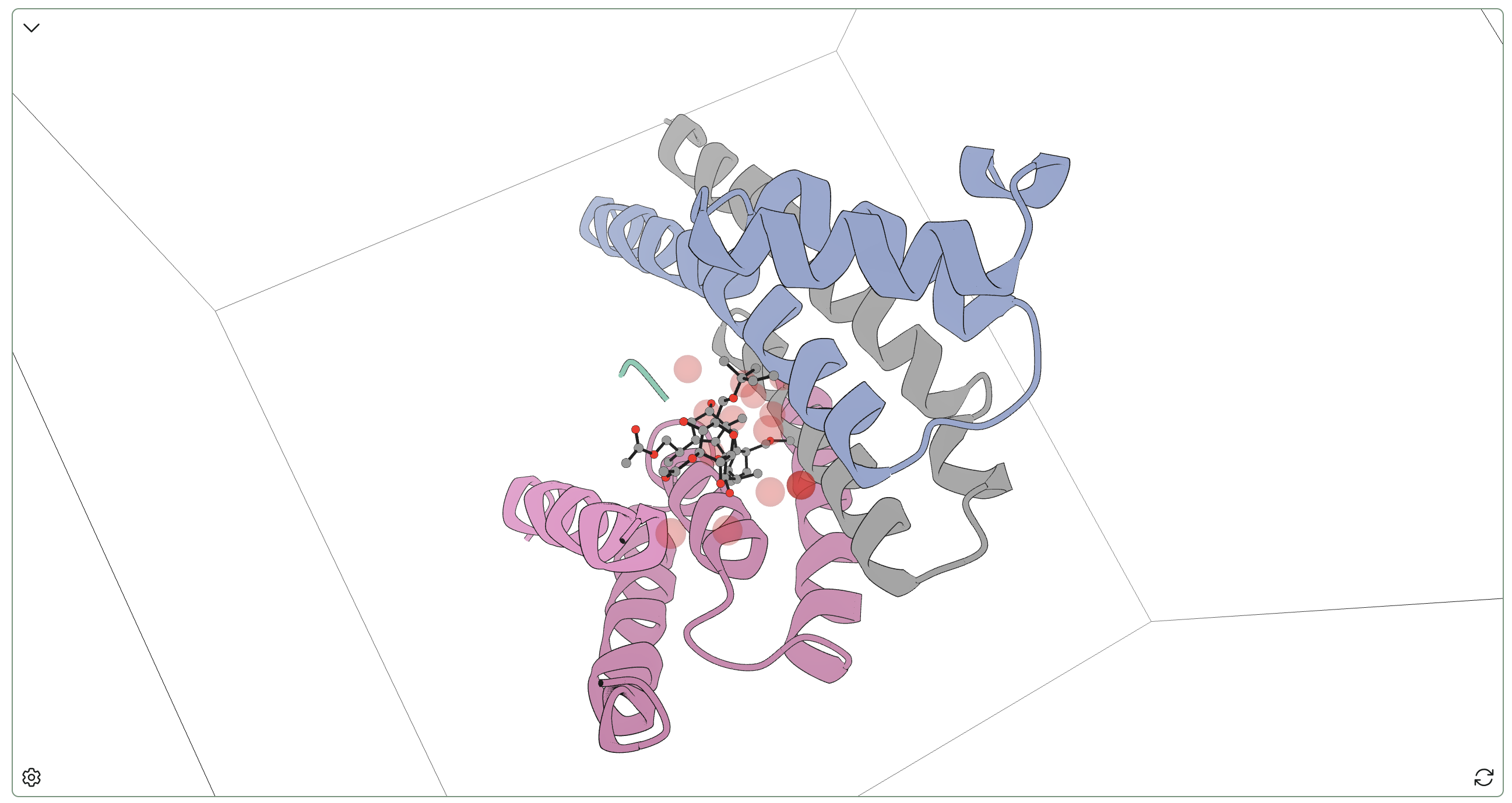
5MXO: 14-3-3 sigma and p53 C-terminal 12-mer complex stabilized by fusicoccin A
The interaction between p53 and 14-3-3 proteins is important in mammalian cell-cycle regulation, and small molecules such as fusicoccin A can stabilize model p53–14-3-3 complexes. Fusicoccin A, a fungal phytotoxin, is particularly interesting as a naturally occurring molecular glue that stabilizes a variety of 14-3-3-containing complexes.
A complex of 14-3-3 sigma, p53 (represented by a C-terminal synthetic 12-mer), and fusicoccin A was originally reported by Doveston and co-workers in 2017, and we examined this complex with Rowan's hydration-site analysis. In contrast to the rapamycin complex above, we found three hydration sites with high protein–ligand bridging occupancy:
- One site connected the hydroxyl on the 5-membered ring of fusicoccin A to SER50.
- Another site connected ASN47 to the pendent crotyl ether of fusicoccin A.
- The last highly bridging site connected ASN47 to the hydroxyl of the central 8-membered ring of fusicoccin A.
Interestingly, there's also a highly occupied hydration site near PHE42 with only 4 unique waters, indicating a conserved interaction. Taken together, these data suggest that water binding is important in understanding the 14-3-3–fusicoccin A interface, and implicate SER50 and ASN47 as more important than they might seem in binding.
These case studies show how Rowan's hydration-site-analysis functionality can be useful in a variety of contexts, from simple molecules like benzamidine to complex macrocyclic molecular glues. If you're working in a structurally-enabled lead-optimization campaign, consider using Rowan!









