What to Do with a Pose
by Corin Wagen · July 15, 2026
Congratulations! You've obtained a protein–ligand pose. Maybe you got this pose from crystallography, maybe you ran a high-throughput docking screen and found a hit that you liked, or maybe you just ran co-folding on a ligand of interest. Here's a quick guide to what you can do next in Rowan.
(This post is motivated by the TBXT med-chem hackathon we hosted with muni, where we saw a number of teams generate plausible protein–ligand poses and then just… stop. The content here should all be common sense to drug-discovery professionals.)
Look at the Pose
The first thing you should do with a pose is look at it! A picture is worth a thousand words, and "this pose has a good docking score" is less valuable than actually trying to understand what interactions are being made (if any).
Rowan makes it simple to view poses in 2D or 3D representations:
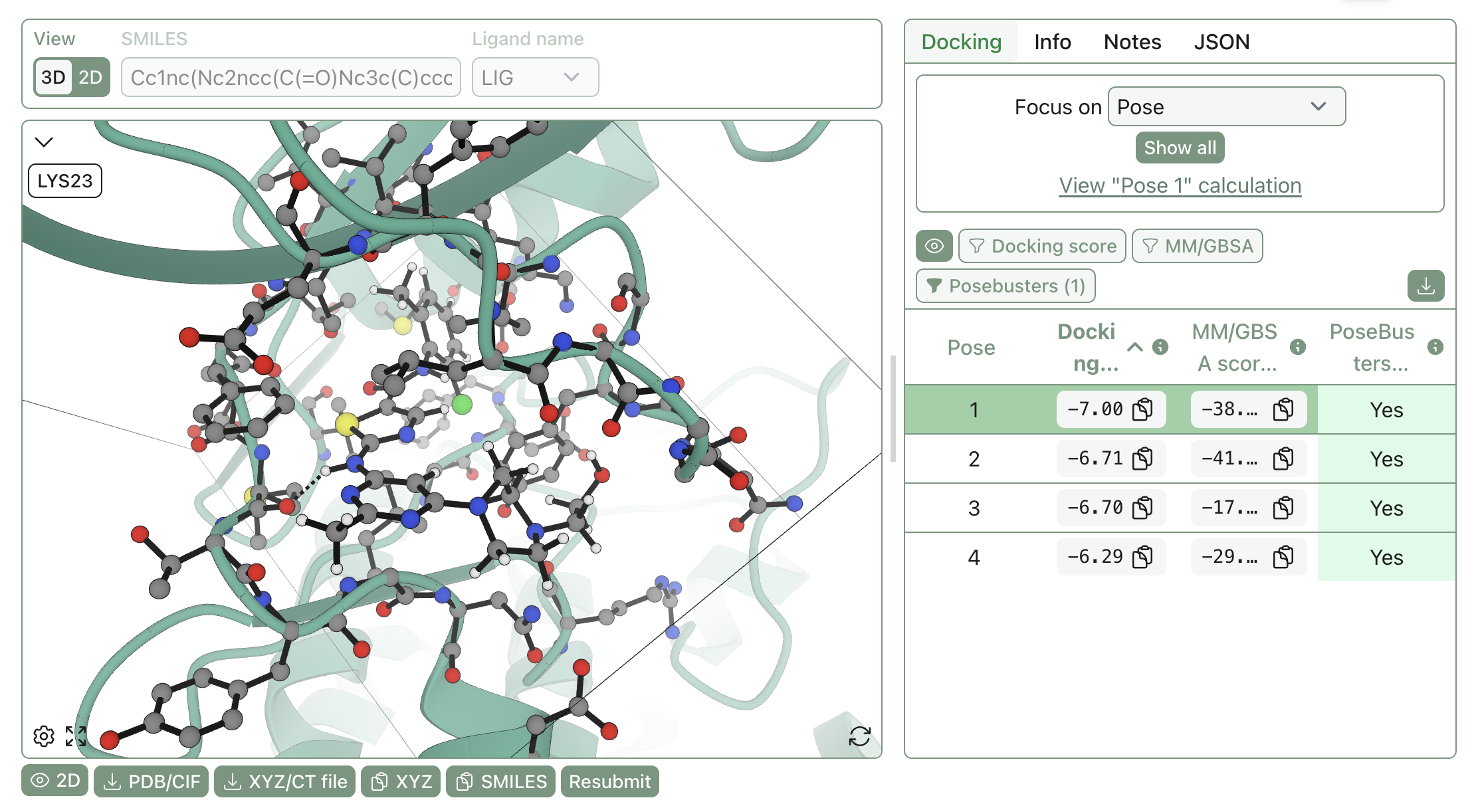
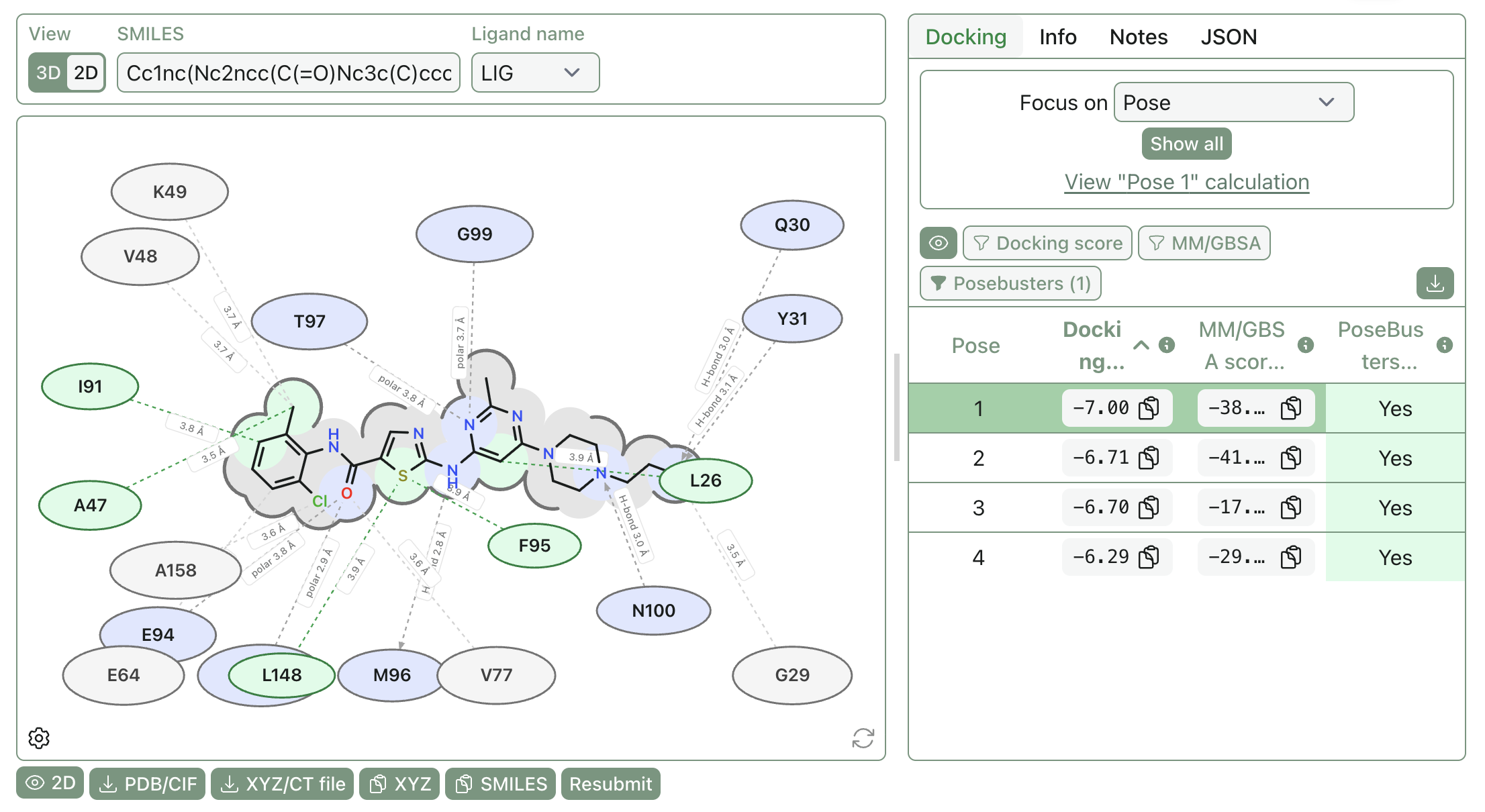
Poses can be superimposed for easy comparison, and proteins can be viewed in surface representation to better illustrate solvent-exposed areas.

Refine and Optimize the Pose
Poses generated by docking and co-folding are often unphysical and contain ligands in highly strained conformations (what some have called "bodies stuffed in a closet"). Optimizing these poses with molecular mechanics is a quick way to reduce the number of unphysical artifacts and generate more realistic geometries. (Plus, this process also adds hydrogens, which are missing by default in many docking and co-folding algorithms.)
If you select "Refine poses" in Rowan's docking or co-folding workflows, Rowan will automatically add hydrogens and optimize the ligand's geometry for you.
Compute the Pose Strain
Optimization removes many of the most heinous errors from docking or co-folding, but some issues can remain. In particular, it's not uncommon to see large and flexible molecules which are predicted to bind in high-energy conformations; while these poses are indeed minima on the potential-energy surface, they're too far above the low-energy solution conformation to be relevant.
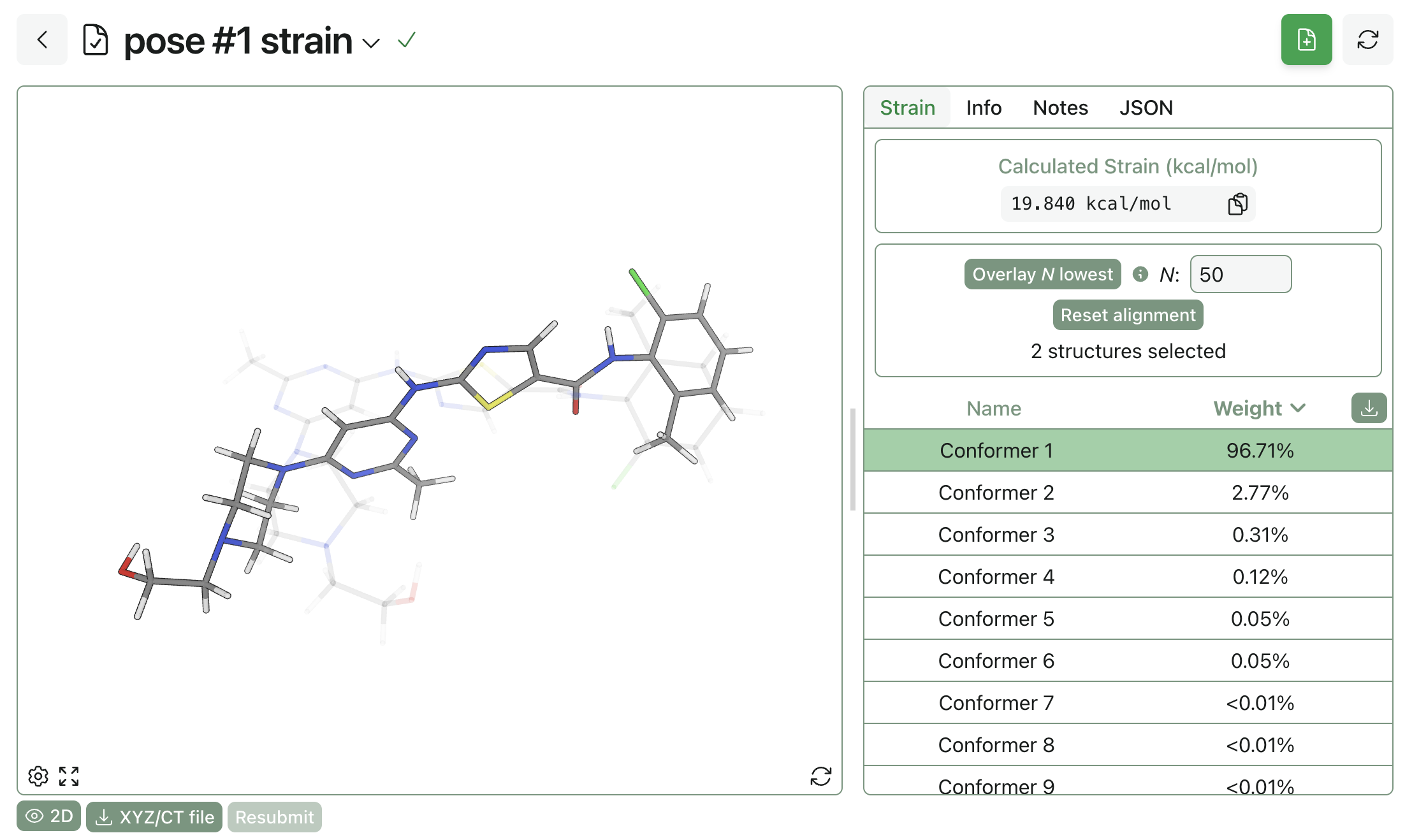
Computing the strain of the docked poses relative to unbound conformers can be a useful way to distinguish reasonable and unreasonable poses (many papers have been written on this topic, see e.g. this work from Sherer and co-workers at Merck). Rowan automatically computes strain when a conformer search is run in docking or when "Compute Strain" is selected in co-folding; strain can additionally be computed separately through Rowan's dedicated strain workflow.
Practically, a strain value of more than 5 kcal/mol is cause for concern, while a strain value of more than 10 kcal/mol should generally be discarded. Running a strain calculation on the "best" docking pose above shows a strain of 19.8 kcal/mol, indicating that this pose is unlikely to be relevant biologically.
Perform Interaction Analysis
Sometimes there are residues that sit near the ligand but don't engage in an obvious salt-bridge or hydrogen-bond contact. These residues can be engaging in an important non-covalent interaction, like a halogen bond or a cation–π interaction, or they can just be sitting there but not doing anything important. In these cases, quantum mechanics–based interaction analysis can be run to figure out the interaction energy associated with individual residue–ligand contacts.
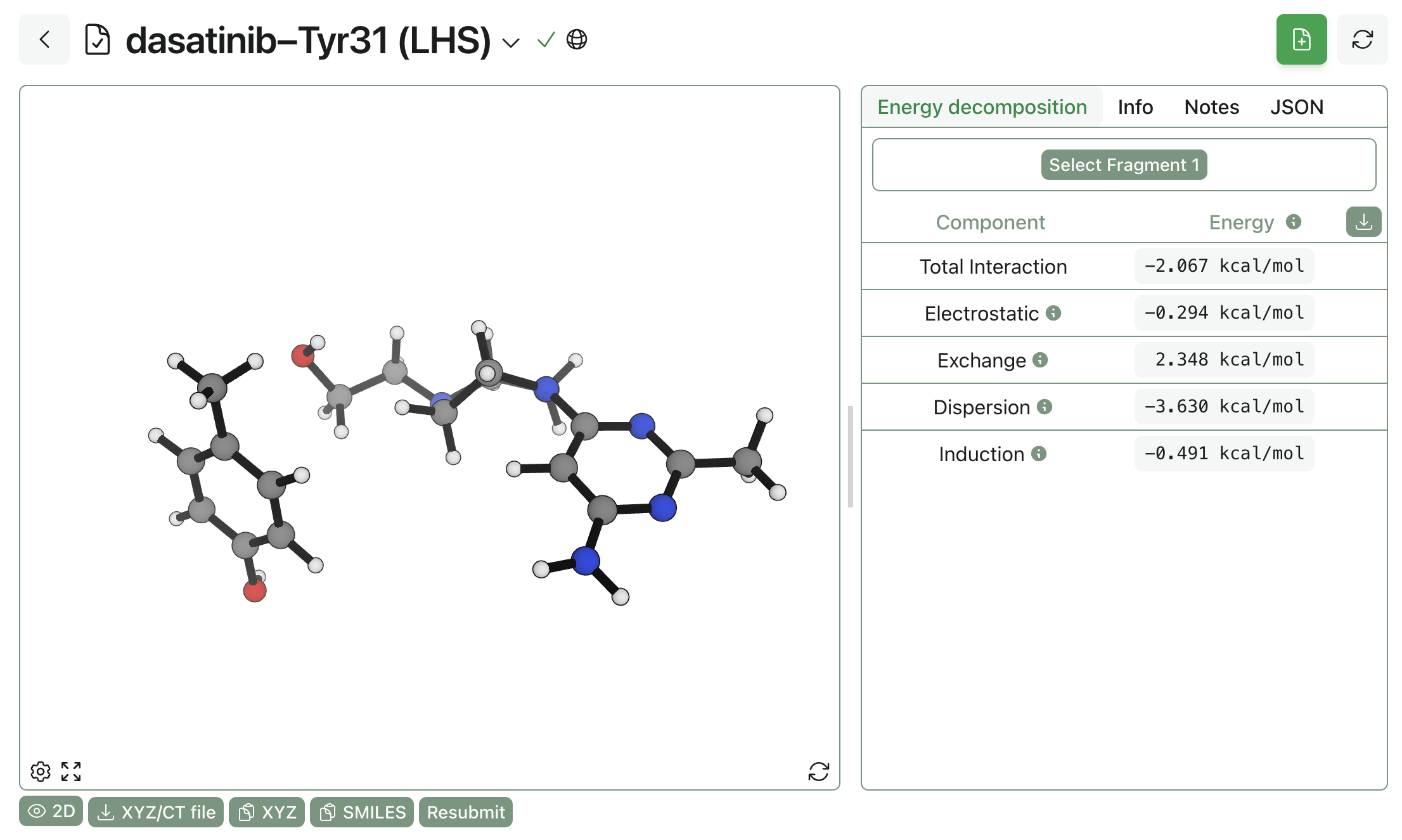
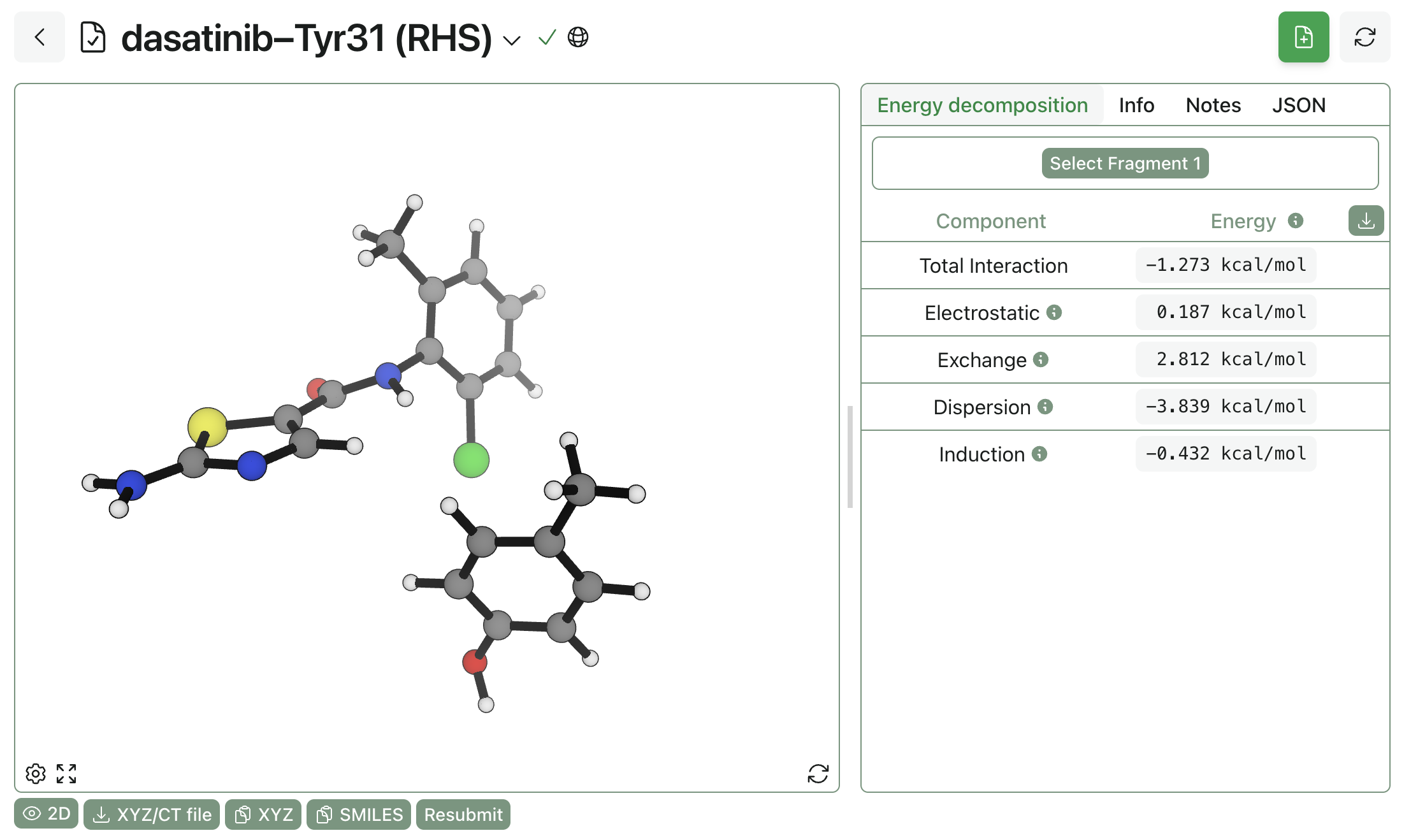
Rowan's interaction-energy-analysis workflow uses symmetry-adapted perturbation theory (SAPT) to decompose the interaction between two fragments into individual quantum-mechanical components. To compute the interaction energy, simply upload the protein–ligand fragments of interest into the workflow, divide them into fragments, and run the workflow. A large interaction energy (i.e. very negative) indicates that the residue is making an important contact, while a small interaction energy indicates that the residue isn't doing much.
Here, the dasatinib pose has been split into two halves to assess how strongly various groups were interacting with a nearby tyrosine residue:
Run Molecular Dynamics
Short molecular-dynamics simulations are a great way to interrogate how a pose behaves in the pocket. MD simulations can be used to study which protein–ligand contacts persist over time, how much the ligand drifts in the pocket, how flexible various receptor sidechains are, and how explicit solvation impacts binding-pose stability.
Rowan's pose-analysis MD workflow automatically parameterizes the ligand, adds solvent and counterions to the protein–ligand system, warms and equilibrates the system, and then runs production MD simulations.
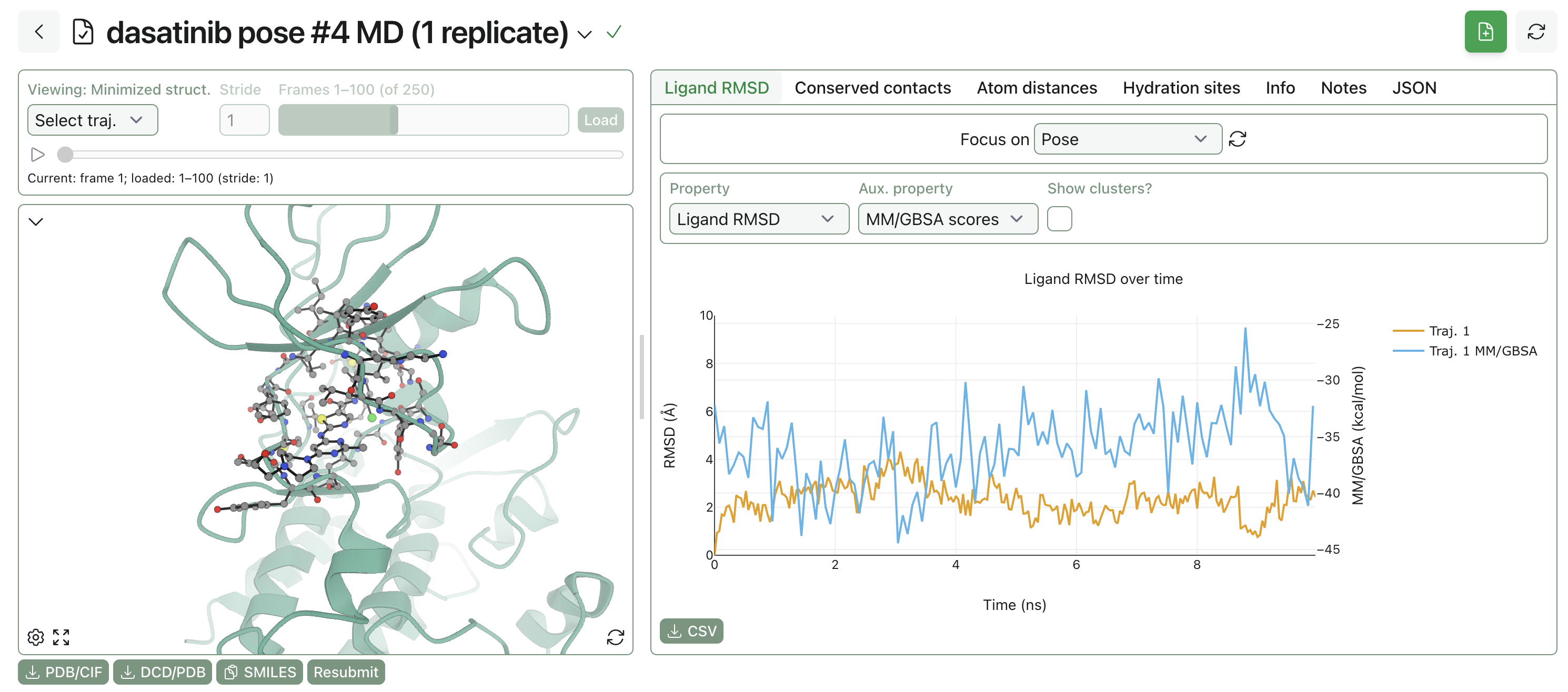
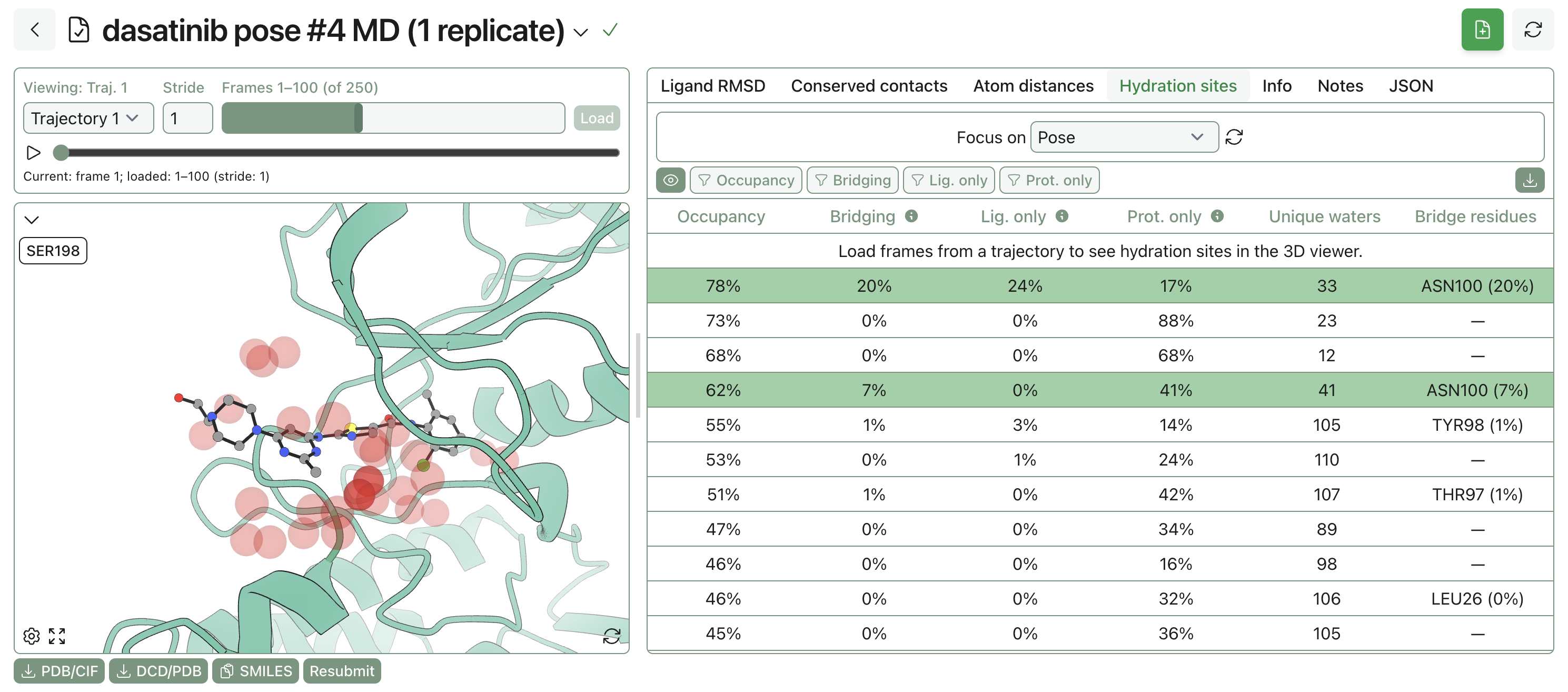
You can look at a variety of parameters once the MD simulation is complete: protein radius of gyration and surface area, ligand RMSD, average MM/GBSA interaction energy, and so on. Rowan also automatically identifies conserved hydration sites in the vicinity of the ligand, which can be used to identify if any seemingly empty spaces in the pocket are actually occupied by structural or bridging waters.
Compute Binding Affinity
If you're trying to design a ligand that binds to a protein, then predicting binding affinity is useful. There are a few low-cost ways to rank protein–ligand binding affinity on arbitrary scaffolds:
- Gnina, a neural-network-based scoring function which can be run through Rowan's docking workflow.
- MM/GBSA, which computes the average molecular mechanics–based interaction energy with generalized-Born implicit-solvent model over an MD trajectory.
- Single-point semiempirical quantum-chemical methods, which use MOPAC or other semiempirical methods to predict single-pose interaction energies with implicit solvent. (This is not yet public; reach out if you want to try these methods in Rowan.)
For optimization along a series, free-energy perturbation (FEP) is the best way to robustly predict binding affinity for compounds. Rowan makes it simple to run all of these methods for compound ranking and prioritization via GUI or API. Read our guide to running FEP here.









