Open-Source Projects We Wish Existed
by Corin Wagen, Jonathon Vandezande, Ari Wagen, and Eli Mann · Sept 9, 2025

The celebration of the peace of Münster, 18 June 1648, in the headquarters of the crossbowmen's civic guard (St George guard), Amsterdam by Bartholomeus van der Helst (1648)
At Rowan, we work to support scientists in many different areas of chemistry: we benchmark existing models and algorithms, write our own code, and try to build robust and accurate solutions for our users and customers.
As a part of this work, we spend a lot of time working with open-source scientific code. We've contributed a bit to open-source software already: Corin maintains pymysym and cctk (among others), Jonathon maintains a scientific cookiecutter for Pixi and steamroll (a package for converting 3D molecular coordinates to RDKit objects), and we released Egret-1, a family of MIT-licensed NNPs, earlier this year. (There are more open-source projects to come!)
Still, we often find ourselves wishing that there was a specific open-source libraries that we could use for some task or another. After a variety of one-on-one conversations with academics and industry scientists interested in contributing to useful scientific software development, we realized that it might be useful to collect this list in a single place.
If you are interested in learning what we think makes for a good open-source scientific project, check out our corresponding blog post with our thoughts: How to Make a Great Open-Source Scientific Project.
Without further ado, here's our list of project ideas—broken down by scientific area.
Quantum Chemistry
"Unbundling DFT"
Corin Wagen
Density-functional theory is very complicated. Historically, every DFT library had to reimplement almost all of the core DFT functionality themselves: from individual ERI calculations all the way up to geometry optimizations and thermochemistry. This "vertically integrated" way of building software is—we feel—more fragile and less sustainable. As these monoliths age, it's difficult to update bits and pieces of these codebases without starting from scratch, which makes building new DFT libraries very difficult.
Projects like libxc (exchange–correlation functionals), libint (one- and two-electron integrals), and libecpint (integrals for effective core potentials) make it much easier to write new DFT implementations. Unfortunately, there are still gaps:
- Building DFT grids, pruning them, and evaluating atomic orbitals over them is still quite tricky.
- Open-source implementations of implicit-solvent models like PCM and CPCM exist, but are buggy and difficult to work with.
- Some of the best low-cost methods use Grimme's gcp counterpoise correction, but working with this correction is tricky. (To our knowledge, no Python interface exists.)
Clean scientific packages addressing each of these tasks would make the actual work of building and maintaining a DFT engine much more manageable.
A Unified Thermochemistry Library
Corin Wagen
The Hessian matrix of nuclear second derivatives can be generated by lots of different methods, but actually converting this matrix into frequencies and thermochemistry values is tricky and easy to get wrong. Lots of programs reimplement this themselves rather poorly—it would be nice to have a standard implementation that automatically removes vibrational and rotational motion, checks for large rotational constants, extracts vibrational frequencies, and returns corrected standard-state thermochemical parameters.
(It's possible we'll build this ourselves at Rowan.)
SCF
Better Electron-Density Guesses
Jonathon Vandezande, Eli Mann
DFT calculations must use SCF to iteratively solve for the electron density, as the potential depends on the orbitals (which depend on the potential). There are many different initial guesses available, such as using the core Hamiltonian, extended Hückel, superposition of atomic density (SAD), fragment orbitals, or projection from a simplified functional or basis set. Various implementations are spread across packages, but few packages implement all of them. ML can also be used to provide a guess density in a fraction of the time. We have generated a large dataset of electron-densities at the ωB97M-D3BJ/def2-TZVPPD level of theory in partnership with Macrocosmos and are happy to work with anyone who is interested in producing an open-source density-guess program.
SCF Convergence
Jonathon Vandezande
SCF convergence is the bane of the existence of computational chemists, randomly killing jobs and arbitrarily converging to high energy states. This is an impediment to large-basis DFT calculations, large-scale DFT workflows, and generation of the data needed for training NNPs. New and faster methods are needed to improve SCF convergence, as resorting to second-order methods is often too expensive. Initial convergence problems can often be avoided with better electron-density guesses. Trailing DFT convergence can often be more insidious, with constant, small energy and density changes preventing convergence. Methods that can detect such trailing convergence and take judicious second-order or ML steps to accelerate convergence will be incredibly useful across a variety of DFT workflows. If this can be implemented in a simple package that is shared across QM codes, the combined experience of many different experts can be used to help tackle this problem.
∆SCF
Jonathon Vandezande
∆SCF (pronounced "delta SCF") is a method for finding excited states of molecules where the electronic density is converged to a saddle point of the electronic Hamiltonian. It is useful for low-lying excitations where orbital relaxation is significant and charge-transfer or core–hole excitations, where TDDFT can often fail. The simple formalism makes it easily amenable to the prediction of the properties of excited states, but it often requires complex SCF convergence methods to ensure convergence to the desired saddle point. This is particularly useful for the modeling of OLEDs, where only a few low-lying excited states are needed (see this article on the effectiveness of ∆SCF for MR-TADF emitters).
A high-quality open-source implementation of various excited-state-aware SCF-convergence methods using the Maximum Overlap Method (MOM), Constrained DFT (cDFT), projection-based methods, optimized orbital selection, or ML-based methods would simplify the process of high-throughput exited-state modeling for systems as diverse as sunscreen candidates, light stabilizers in polymers, and deep-blue OLEDs.
Better Implicit-Solvent Models
Corin Wagen
Implicit solvent is a major source of error in computational chemistry. DFT calculations frequently use solvent models like CPCM, IEF-PCM, or SMD; semiempirical methods and forcefields use simpler Poisson-Boltzmann or generalized-Born methods; and most NNPs don't have any support for solvent. Since future xTB versions may drop support for CPCM-X, the best low-cost solvent model that we're aware of, new solvent models compatible with semiempirical methods or NNPs are badly needed.
Recent work from Sereina Riniker and coworkers (ref, ref) shows that graph neural network–based solvent models can outperform physics-based implicit-solvent models, at least on certain benchmarks. We'd love a simple and modular implicit-solvent model that could be easily combined with other methods—there are many different scientific approaches that one could try here, almost all of which would probably be useful to the field. Ideally, it would be possible to use any sort of computational method with the solvent model, much like Grimme's D3 dispersion correction can be combined with physics-based and ML-based methods.
Advanced Spectroscopy: NMR, ECD, and VCD
Corin Wagen
To our knowledge, there's no good open-source implementation of many complex spectroscopic properties. Predicting nuclear magnetic resonance (NMR) spectroscopy, electronic circular dichroism (ECD), and vibrational circular dichroism (VCD) is a common task in the pharmaceutical industry, but today's open-source packages can't predict these properties. This is probably too much for a single standalone package—but if you're developing open-source DFT software, consider including some spectroscopic methods!
Optimization and Reactivity
Implicit Coordinate Conversions
Corin Wagen
In theory, optimizing molecular geometries isn't too hard: simply use your quantum chemical method of choice to get nuclear gradients and use an established algorithm like L-BFGS to find the nearest minimum. In practice, direct optimization in Cartesian coordinates is pretty inefficient. Molecular systems tend to have highly coupled gradients, and converting to "internal coordinates" like bond lengths, angles, and dihedrals can often reduce the number of gradient calls needed by about an order of magnitude.
Converting from Cartesian coordinates to internal coordinates and back gets a little complex. While lots of quantum-chemistry programs or optimization libraries have their own internal-coordinate implementations, we're not aware of any robust standalone utilities that implement this conversion. This would be very useful!
Better Optimizers
Ari Wagen
Optimizing a structure with density–functional theory (DFT) is often the most time-consuming and compute-intensive step in molecular modeling workflows, and low-cost methods like neural network potentials (NNPs) promise to speed these workflows up. However, the best optimizers from the DFT era don't always perform reliably when paired with new low-cost methods.
It'd be super helpful to have a lightweight optimizer package that can reliably find local minima on a potential energy surface in as little wall clock time as possible. Because NNPs and semiempirical methods like xTB are so fast, it's not uncommon for the time-limiting factor in an optimization to be the optimizer itself, meaning that the fastest reliable optimizers may deliberately take more, smaller steps to keep each step's cost down.
Metadynamics-Based Conformer Search
Jonathon Vandezande
Current conformer search methods are often either stochastic searches or metadynamics-based. The former are incredibly fast, while the latter are more robust. CREST is currently the best metadynamics-based conformer search code, but due to its tight integration with xTB (and being written in Fortran), it is not amenable to use with NNPs.
It would be useful to have a Python package that can orchestrate batches of metadynamics runs on a single GPU to perform conformer searches with NNPs using iMTD-GC and iMTD-sMTD or variants thereof. Features should include:
- Conformer screening
- Constraints
- Non-covalent complex sampling
Standalone Conformer Deduplication
Corin Wagen
There are many methods for generating conformer ensembles: RDKit, CREST, Omega, MacroModel, MD simulations, diffusion and flow-matching methods, and more. Unfortunately, there are many fewer ways to filter and deduplicate conformer ensembles. Various packages like CREST offer ways to do this as a part of their software, but it's not always possible to easily combine different conformer-generation methods with deduplication.
A modular and tunable conformer-deduplication library would be very useful and significantly simplify using modern ML-based conformer-generation methods.
Transition States
Variational Transition-State Theory
Corin Wagen
Variational transition-state theory (VTST) is a beautiful way to circumvent some of the key assumptions of transition-state theory. Briefly, VTST extends the conventional idea of the transition state to encompass the entire dividing hyperplane normal to the intrinsic reaction coordinate and optimizes the hyperplane to find the position where the "flux" (reaction rate through the hyperplane) is smallest. (This is a cartoon overview of VTST—for a more complex explanation, see this review by Truhlar and coworkers or any number of other reviews.)
In cases where recrossing limits the applicability of a single transition state or the entire reaction surface is "loose" (like many radical reactions), VTST can be significantly more accurate. A big limitation of VTST is speed, though. Since VTST takes many more gradient/Hessian evaluations than regular transition-state theory, only small systems can be studied with conventional quantum chemical methods like DFT.
The canonical VTST package is POLYRATE, which, while groundbreaking, is quite difficult to work with. (For instance, integrating with Gaussian takes an entirely separate package, GAUSSRATE.) We would love to experiment with using VTST in combination with low-cost methods like NNPs, but trying to plug an ML model into POLYRATE just seems hopeless. A clean Pythonic rewrite of POLYRATE would make it possible to explore using NNPs to scale VTST to larger systems.
Preparing Structures for Double-Ended TS Search
Jonathon Vandezande
Double-ended TS search methods such as the freezing-string method (FSM) and nudged elastic band (NEB) perform best when the ends are placed in favorable positions in the reaction channel. Placement of the active species far from each other or in highly strained positions can cause the TS search to be slow or even fail. Fully automated TS-finding methods need smarter initial placement of the reactants and products; manual determination of the correct atom indexing or hand-placement of the initial structures cannot scale to thousands of reactions.
Simple Python packages would be very helpful for:
- Determination of corresponding atoms between reactants and products
- Initial placement of structures in the reaction channel
Nudged Elastic Band
Jonathon Vandezande
NEB and FSM are the two major competing methods for double-ended TS-search methods. FSM tries to climb the channel from both sides, while NEB attempts to stretch a band from the initial interpolation down to the reaction channel. Having multiple methods to find transition states allows for more robust TS searches, and pairing of methods may lead to easier determination of reaction paths. Easy integration of NEB with NNPs will accelerate these calculations, and help make automated TS search much more routine.
Single-Ended GSM
Jonathon Vandezande
While double-ended TS search methods are useful when the desired products are known. It can often be useful to explore possible chemical reactions without knowing a priori which ones will actually happen. Similar to ML-FSM for double-ended TS search, a package for single-ended GSM should support:
- Freezing string method (FSM)
- Multiple interpolation methods
- Reaction-channel vectors for initialization
- Efficient optimization
- Easy callbacks for saving/inspection of intermediate states
Cheminformatics
Robust 3D ⇒ 2D Conversion
Corin Wagen
In computational chemistry, there's a divide between "3D" representations of molecules, which contain information about the position of atoms in space, and "2D" representations, which do not:
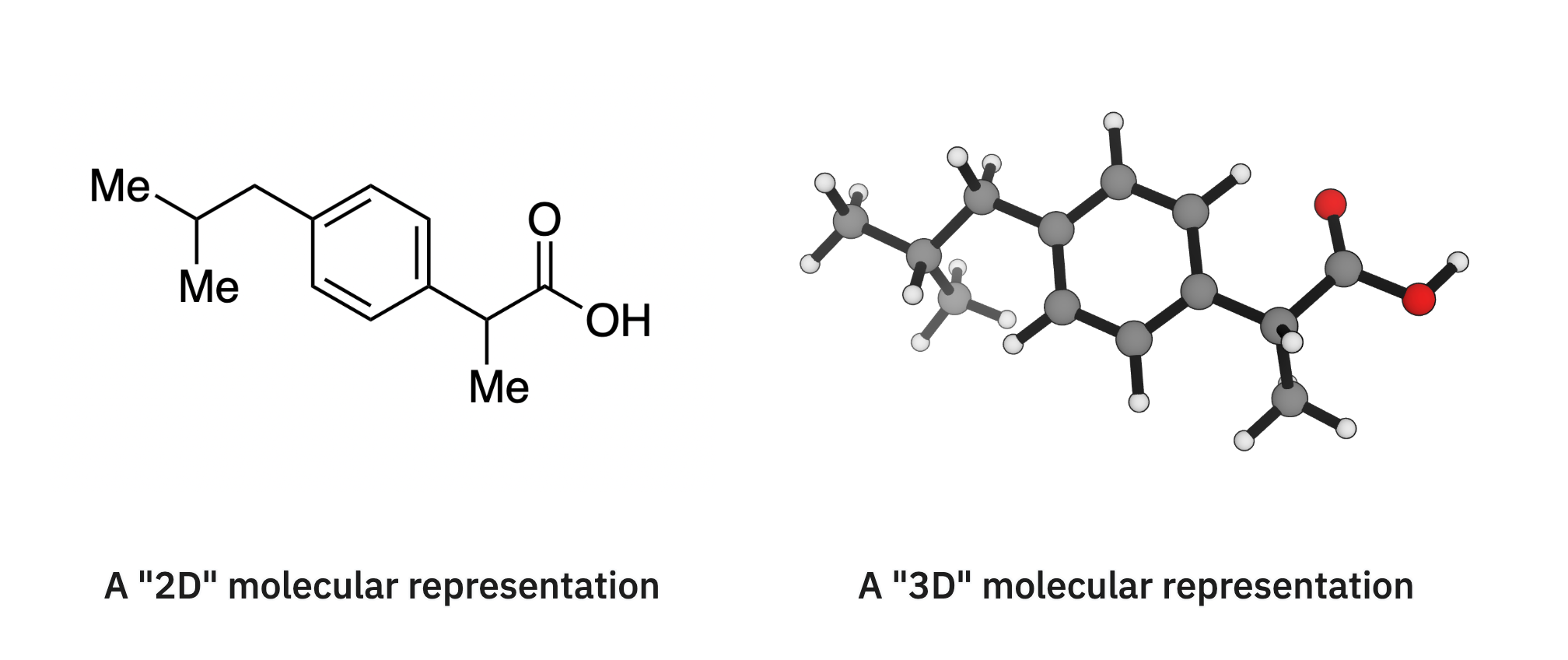
Converting from 2D to 3D is a well-studied problem: this is essentially just a conformer search, and algorithms like ETDKG work well for most cases. In contrast, converting from 3D to 2D is difficult and error-prone. We've done a little work in this area by creating "steamroll," a package which packages and wraps Jan Jensen's xyz2mol code and automatically tries different settings, but better algorithms are badly needed here.
Core RDKit Functions in Rust
Ari Wagen
The RDKit is an incredibly powerful program supporting countless computer-aided drug design teams and academic labs. The RDKit is written in Python, with core functions written in C++ for performance.
As the cost of compute scales, we anticipate demand for core RDKit-style functionality increasing. We'd love to see core functions from the RDKit carefully rewritten and optimized in Rust, a modern memory-safe language built for speed.
We envision this being done in a completely RDKit-compatible way so teams can benefit from the compute optimization without changing their workflows. Core functionality includes:
- SMILES to 2D representation
- 2D representation to standardized, truly "canonical" SMILES
- ETKDG-like 2D to 3D inflation
- Tautomer enumeration and standardization
- Basic graph-based descriptor calculations (ex. AMW and num. rotatable bonds)
- Substructure search
- Chemical similarity search
Butina Splitting and Overlap Checking
Eli Mann
Any machine learning project that uses molecular data should use a more robust data splitting method than a random split. When random splitting, structurally similar molecules may appear in both train and test sets, leading to data leakage and inflated performance metrics.
This issue has been investigated before: in a blog post, Pat Walters explores different methods for splitting chemical datasets and ultimately recommends Butina splitting on Morgan fingerprints. This splitting technique groups molecules by their Tanimoto similarity of Morgan fingerprints, then assigns different groups to train, validation and test sets to avoid data leakage of structurally similar molecules. We've consistently observed dramatic performance drops on validation/test sets when we switched from random to Butina splitting. For example, in our aqueous solubility work, random splitting yielded a validation R2 of 0.9 while Butina splitting yielded an R2 of 0.75.
Pat provides an RDKit-based implementation of Butina clustering on Morgan fingerprints. While fast, this method is extremely memory-intensive since due to the O(N2) pairwise Tanimoto similarity calculations. To compute the Butina split of the 304k molecule GEOM-DRUGS dataset, I needed to spin up a CPU instance with multiple TB of memory.
The open-source package should contain the following:
- A fast, memory intensive method for Butina clustering small datasets (like Pat's RDKit implementation).
- A slower, constant memory Butina clustering method (or approximate method) for larger datasets.
- Train/validation/test set splitting using the Butina clusters.
- Tanimoto-similarity overlap checking between datasets to determine data leakage for external benchmark datasets.
A first iteration should compute Tanimoto similarity on Morgan fingerprints, with support for other descriptor methods in future releases. Other splitting methods like Murcko scaffold splitting or simpler molecular backbone overlap detection would be nice additions to this project. With minimal dependencies, this could become the standard package for molecular dataset splitting.
Conclusion
This is a non-exhaustive list—we haven't discussed any protein-related utilities (and there's certainly lots more that could be done there). We may add to this list or create new lists in the future.
If you're interested on working on any of these, please reach out! We'd love to talk more and help point anyone in the direction of what we think would be useful for the field. And if you decide to take on one of these challenges, please take a look at our notes on good scientific software development.










