Hydrogen-Bond-Basicity Predictions for Scaffold Hopping in PDE2A Inhibitors
by Corin Wagen · Feb 14, 2025
In a 2018 study (J. Med. Chem. 2018, 61, 1001–1018), Chris Helal and co-workers at Pfizer reported the development of phosphodiesterase 2A (PDE2A) inhibitors as potential treatments for cognitive disorders in schizophrenia. Their core scaffold—pyrazolopyrimidine 1—exhibited good potency but remained relatively lipophilic, leading to high human-liver-microsome clearance and thus an excessively high estimated dose.
The authors investigated peripheral modifications to decrease lipophilicity, but found that the perturbations they tried either compromised activity or led to increased values of MDR BA/AB. (MDR BA/AB is the ratio of basolateral-to-apical flux to apical-to-basolateral flux of a compound in MDCK cells engineered to express P-glycoprotein (MDR1), reflecting P-gp–mediated efflux). Accordingly, the Pfizer team next investigated re-engineering the central heterocyclic core to preserve potency while improving lipophilicity and efflux.
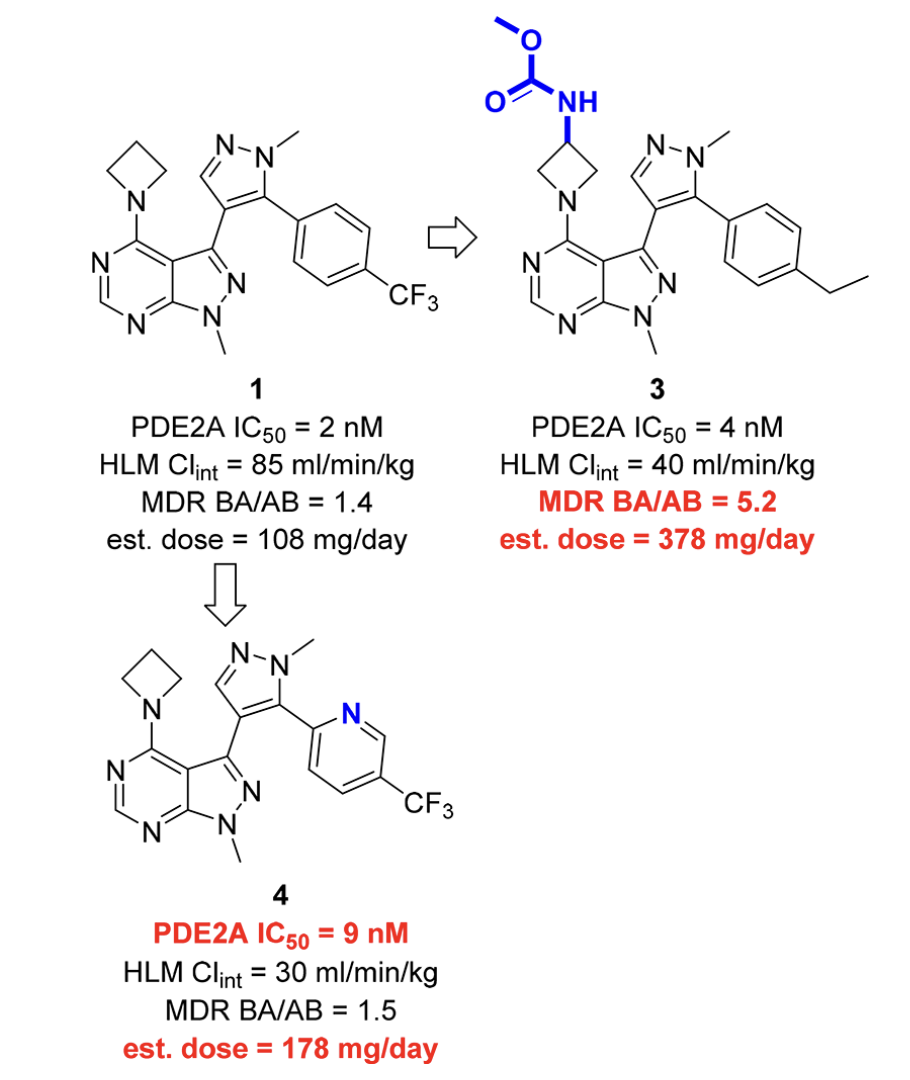
Peripheral modifications to 1 were unsuccessful.
LMP2 Calculations Identify a More Potent Scaffold
Inspired by results from Bayer showing that different heterocyclic isomers could lead to 50x potency improvements for PDE5A, the team explored an imidazotriazine ring to replace the pyrazolopyrimidine scaffold. Counterpoise-corrected LMP2/cc-pVTZ//X3LYP/6-31G** calculations in Jaguar indicated that key hydrogen-bond interactions in the enzyme's active site would be strengthened with the imidazotriazine core. This prediction was borne out experimentally: after extensive optimization, the new scaffold led to the clinical candidate PF-05180999, which demonstrated higher PDE2A affinity and improved brain penetration.
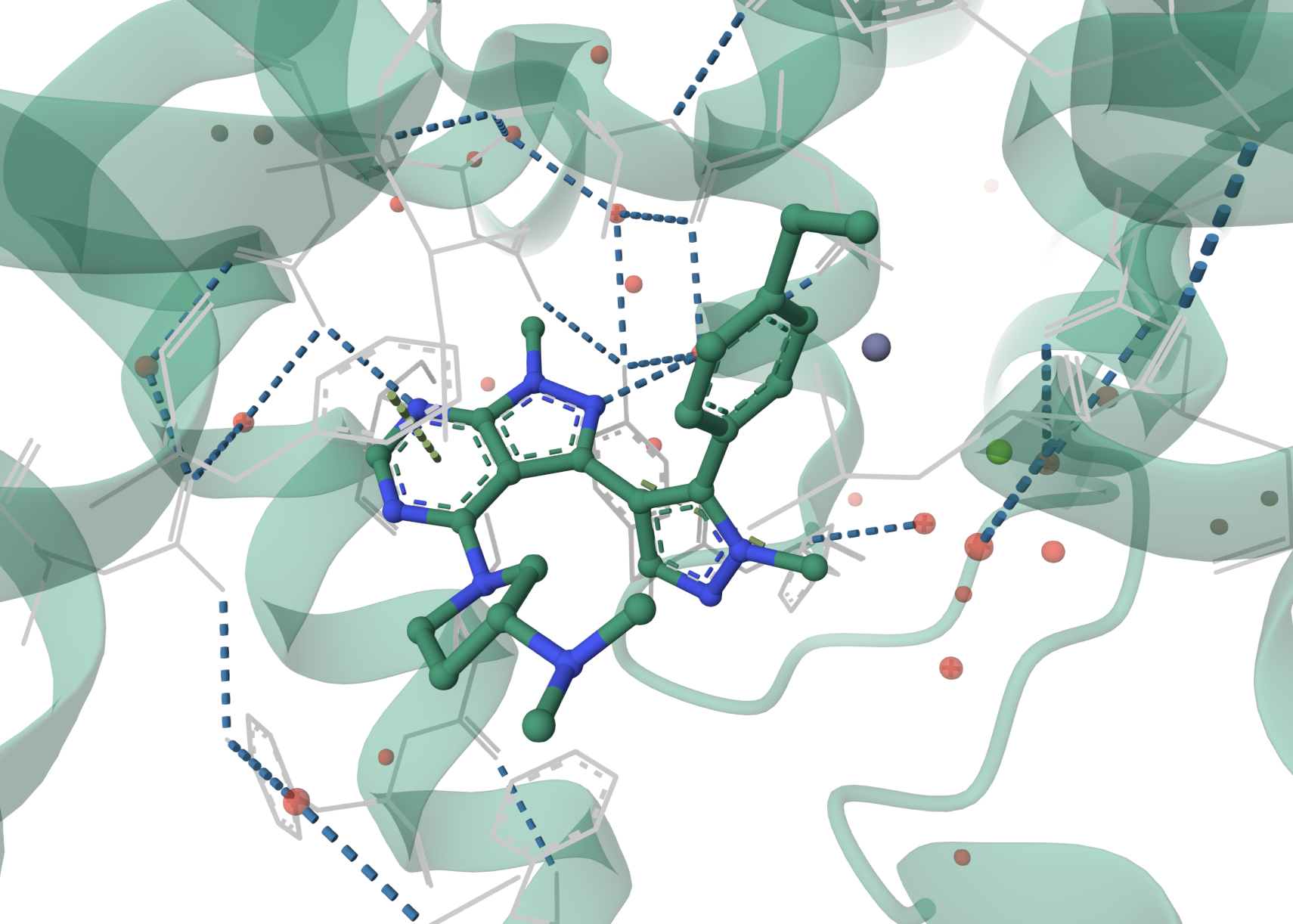
Illustration of the inhibitor class bound to PDE2A and the key hydrogen bonds made with the protein (PDB 5UZL).
pKBHX Workflow as a Black-Box Alternative
Unfortunately, the LMP2-based calculations run by Helal and co-workers are challenging for non-expert practitioners to perform accurately: proper setup of these calculations requires properly aligning new ligands with experimental or predicted protein–ligand complexes, deleting all but the relevant regions of the protein, and then hoping that the generated structure is both small enough to enable practical MP2 calculations and large enough to capture all relevant interactions.
Rowan's hydrogen-bond-basicity-prediction workflow (pKBHX) offers a more accessible way to estimate site-specific hydrogen-bond acceptor strengths. Whereas LMP2-based workflows require specialized resources and expertise, the pKBHX approach uses only a single density-functional-theory calculation per molecule, automatically handles different conformers, and has been calibrated and validated against a large experimental database. This method identifies the most strongly hydrogen-bonding heteroatoms, enabling medicinal chemists to prioritize promising scaffold hops without advanced quantum-chemistry infrastructure.
Agreement with High-Level QM Calculations
In an independent analysis of the same scaffold hop, pKBHX calculations confirmed that the imidazotriazine ring would generally strengthen the critical hydrogen bond to the five-membered ring (N2 when using the pyrazolopyrimidine numbering) compared to the original pyrazolopyrimidine core: Rowan predicts an increase in pKBHX of 0.88 units (almost an order of magnitude), while Helal and co-workers predict that the hydrogen bond will be 1.4 kcal/mol stronger.
Both the LMP2-based computations and the pKBHX results also agree qualitatively on the overall increase in hydrogen-bond strengths upon scaffold modification. The two methods disagree on the effect of the scaffold hop on N5, suggesting that in this case side-chain and water-mediated interactions can slightly alter outcomes for ligand-only predictions. Nonetheless, the overall conclusion is the same: substituting imidazotriazine yields a net improvement in hydrogen bonding and potency.
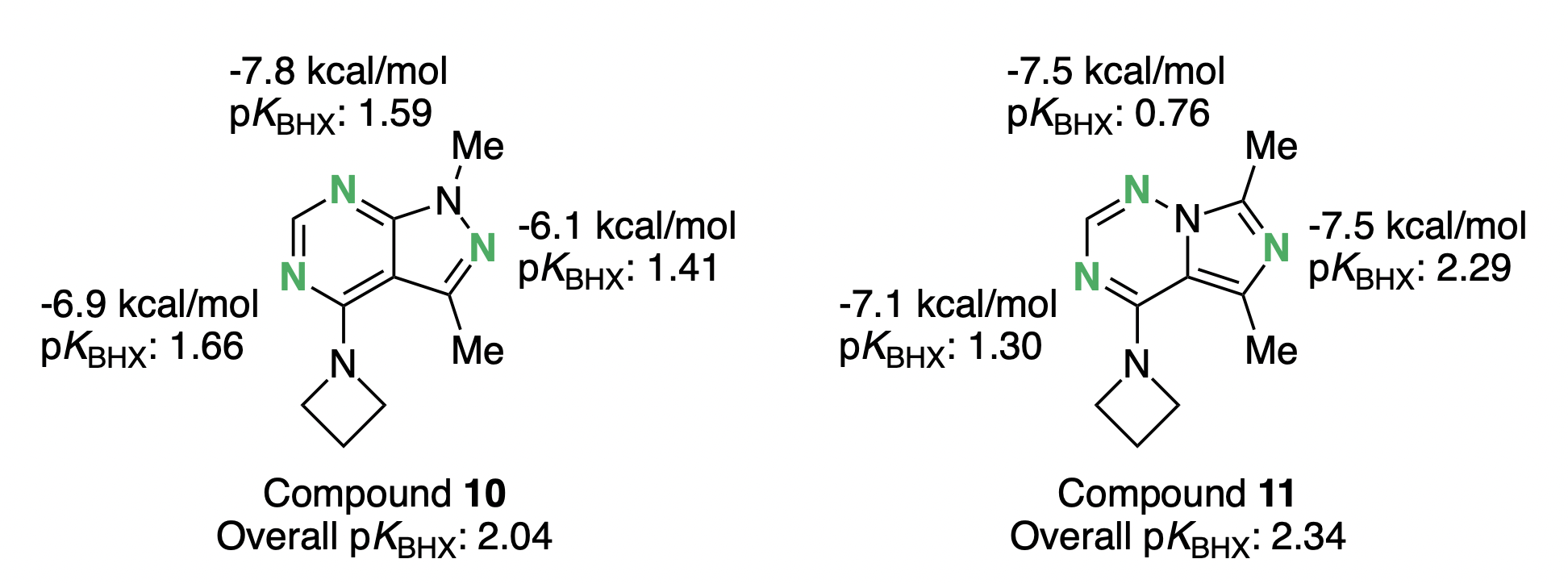
Comparison of the hydrogen-bond-acceptor strength of the pyrazolipyrimidine 10 and the imidazotriazine 11. More negative LMP2 values imply stronger hydrogen bonding; higher pKBHX values indicate stronger hydrogen-bond acceptors.
This case study demonstrates how modeling and simulation tools like the pKBHX workflow can help make complex decisions like scaffold hops more data-driven, accelerating project timelines and decreasing the role of luck in hit-to-lead optimization. While top pharmaceutical companies like Pfizer have long relied on sophisticated computational workflows to help make these high-stakes decisions (as shown by this paper), it's now possible for even small teams to take advantage of these insights.
Rowan is working to democratize computational chemistry and bring advanced design & simulation tools to every scientist, not just computational experts. If you're interested in working with us, reach out to our team at contact@rowansci.com or start using our web-based computational chemistry platform today!










