Rowan Research Spotlight: Ameer Nizami
by Corin Wagen · Mar 25, 2025
In the race to build a better battery, few elements look as enticing—or as challenging—as sulfur. It's cheap, abundant, and theoretically can store more energy per gram than traditional lithium-ion materials. Yet making a practical lithium–sulfur (Li–S) battery is notoriously difficult. Figuring out how to build such a battery is the focus of Ameer Nizami, a third-year PhD candidate in the chemical and materials engineering department at Concordia University.

Ameer Nizami
"I am a hybrid researcher in that I do experimentation as well as theoretical modeling," Ameer said during a conversation with Rowan's Corin Wagen. By bridging lab work and computational chemistry, he hopes to solve Li–S's notorious challenges—particularly the "polysulfide shuttle effect" that erodes battery life and performance. If successful, Li–S batteries could one day deliver more capacity at lower cost, paving the way for applications ranging from grid storage to electric vehicles.
Challenges and Opportunities For Lithium–Sulfur Batteries
Li–S batteries look great on paper. Sulfur is cheap, abundant, and boasts a significantly higher theoretical capacity than today's lithium-ion materials (1650 mAh/g, almost an order of magnitude higher than conventional lithium-ion batteries). But this chemistry also has three main drawbacks: sulfur's 80% volume expansion and contraction during cycling, its limited conductivity, and the formation of soluble "polysulfides" that can corrode the anode of the cell. The Li–S redox cycle proceeds from S8 to Li2S via Li2S8, Li2S6, Li2S4, & Li2S2, and each of these polysulfide intermediates can potentially cause chaos and cell degradation.
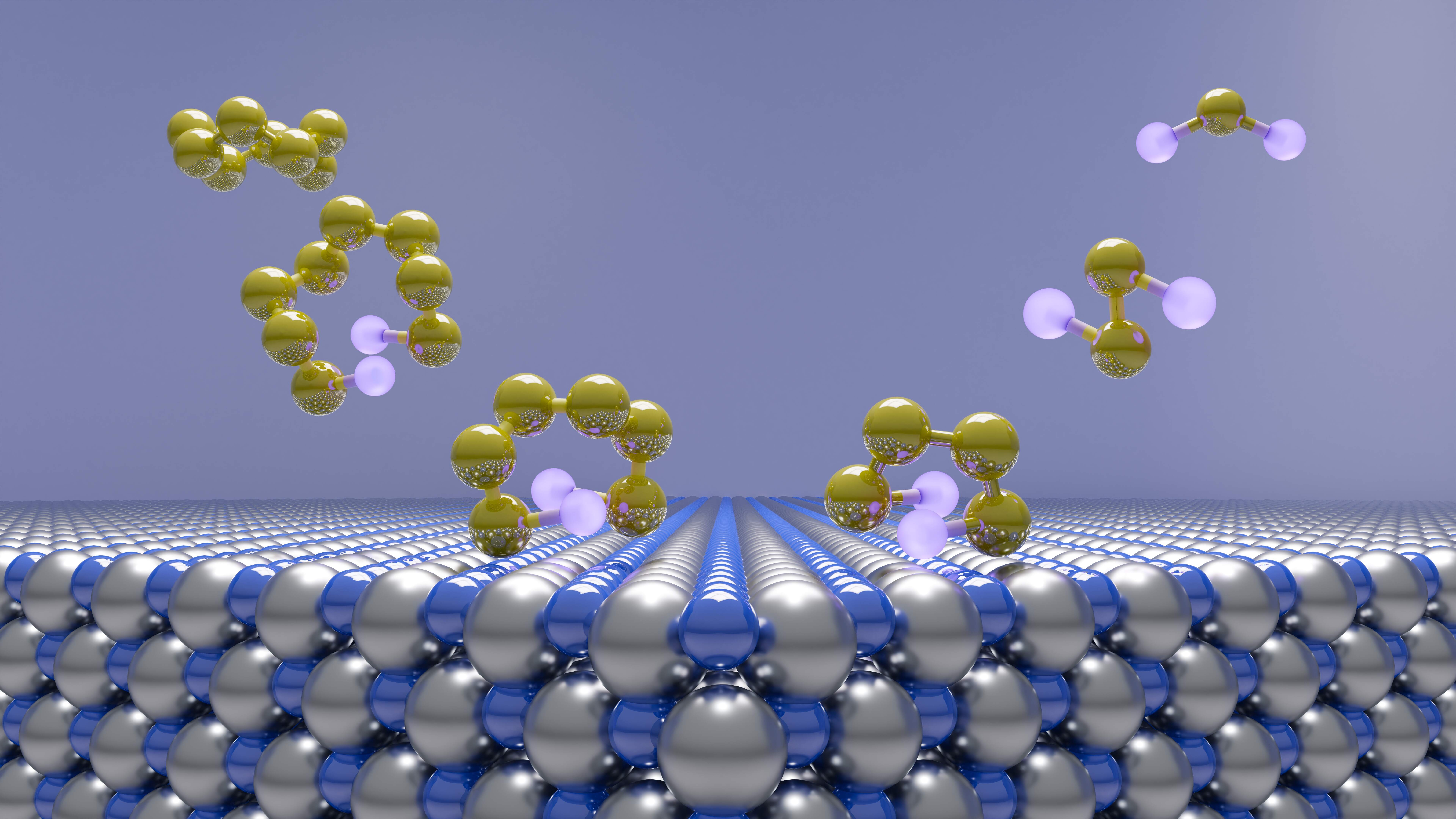
An illustration of different sulfide species relevant to Li–S batteries.
At Concordia, Ameer is tackling Li–S challenges via two main strategies: atomic layer deposition (ALD) of inorganic coatings and the development of specialized polymer films. ALD allows an ultrathin layer of metal or metal nitride to coat the sulfur cathode, improving its durability and helping trap polysulfides. Meanwhile, polymer scientists in his lab formulate "sticky" coatings to hold sulfur in place, increase conductivity, or even self-heal minor damage over multiple cycles. These polymer coatings can dramatically improve the longevity of Li–S batteries, addressing one of the key real-world limitations preventing broader adoption.
But there's a lot of guesswork when designing those new materials and figuring out what they're doing—at least without good computational tools. That's where Ameer's dual expertise comes into play. Working closely with experimental scientists in the lab (or his own experiments), he runs theoretical simulations to see how well new coatings or catalysts might bind polysulfides. These calculations can be demanding and slow, requiring expensive DFT calculations with large basis sets, but provide valuable insights that can guide future research.
Rowan's Computational Platform
According to Ameer, Rowan has made computational chemistry easier and far more intuitive. "When I saw Rowan I said this is modern, this is a sleek GUI interface, this is crazy," he recalled, describing how the platform let him quickly build and optimize molecular structures without the pain of configuring a high-performance computing cluster or manually specifying basis-set files. Now Ameer uses Rowan's low-cost composite methods to explore geometry optimizations and generate plausible configurations for sulfide-bound battery materials.
Rowan makes dealing with complex computational workflows much easier for Ameer. "I tried installing xtb the other day like you guys use. I run it, the kernel breaks. I don't know, I've never had a kernel just die on me… it's a pain," he recounted. Cloud-based platforms like Rowan that don't require users to configure their own compute resources can substantially reduce the overhead required to run computational simulations for Ameer's projects, letting him focus on the scientific tasks at hand.
The Future of Materials Science
Moving forward, Ameer expects that more researchers will become hybrid computation/experimental researchers as tools like Rowan lower the barrier to running simulations. Better software means "it won't take a whole computer science degree on the side" to run calculations, he says, "so I do think that the incoming students will more and more adopt some of these AI tools." He points to recent research from MIT that shows a dramatic uptick in patents and discoveries among researchers that used AI tools, indicating that real productivity gains are possible through the use of these tools.
Ameer also sees a role for computational platforms in curating the best AI tools for researchers who themselves might not be experts in theory or simulation. He describes learning about a new solubility prediction model only through Rowan's social media: "I don't think I would have ever seen it had it not been added by Rowan," he says, "I don't get to read all these papers… there's too much." This year, Ameer has organized a series of workshops on "AI for Materials and Molecules," helping to share resources and information with other scientists in Canada and promote innovation at the intersection of machine learning and materials science.
Moving forward, Ameer expects that Li–S batteries can become key for grid-scale energy storage, where their dramatically increased capacity will be crucial in scaling renewable energy sources to meet demand. He plans to continue integrating computational tools like solubility prediction, NNPs, and more into his workflow, helping him discover new battery materials more efficiently and drive innovation in this exciting space. Stay tuned for more exciting publications coming soon!
If you're interested in following along with this research, you can follow Ameer on LinkedIn.
And if you're interested in being the subject of a research spotlight like this, fill out our interest form and we'll get back to you!










