Introducing Egret-1
by Eli Mann, Corin Wagen, Jonathon Vandezande, Ari Wagen, and Spencer Schneider · Apr 30, 2025
Today, we're releasing Egret-1, a family of open-source NNPs for bioorganic simulation.
Over the past two years of building Rowan, we've probably talked to over a thousand chemists about how they wish they could be using computation. One theme that stands out is that scientists want to be able to trust their results. Historically, there's been no good way to quickly run simulations that weren't largely incorrect—low-cost computational methods have been around forever, but they often behave more like random-number generators than trusted scientific tools.
In theory, neural network potentials (NNPs) can enable fast and accurate chemical simulation, but previous generations of NNPs haven't been as reliable as the legacy quantum-mechanics-based algorithms that chemists are used to—so the simulations are very fast, but chemists still have to double-check the results with quantum chemistry.
Egret-1 changes this. On many relevant benchmarks, our models match or exceed the accuracy of quantum-mechanics-based simulations while running orders-of-magnitude faster. With Egret-1, scientists can quickly get trustworthy results from computation to guide their work.
Training on purpose-built datasets
The Egret-1 family of NNPs comprises three pretrained models built for different use cases:
- Egret-1. Egret-1, a general-purpose model for bioorganic simulation, was trained on 951,005 structures of 19,228 unique organic molecules containing the elements C, H, N, O, F, P, S, Cl, Br, and I. Egret-1 equals or exceeds the accuracy of quantum chemistry for many simulation tasks.
- Egret-1e. Egret-1e ("e" for energy) is built for thermochemistry. It was trained on the same data as Egret-1 plus an additional 784,874 equilibrium organic and inorganic small molecule structures, including nearly 80,000 new unique molecules (and adding support for silicon). This additional data makes Egret-1e excellent at energy predictions, but the lack of non-equilibrium structures makes the model worse at gradients and frequencies.
- Egret-1t. Egret-1t ("t" for transition states) is built for reaction modeling. It was trained on the same data as Egret-1 plus an additional 128,167 structures of neutral transition states, reactive structures, and cycloaddition reaction profiles, improving the model's performance on reactivity-related tasks.
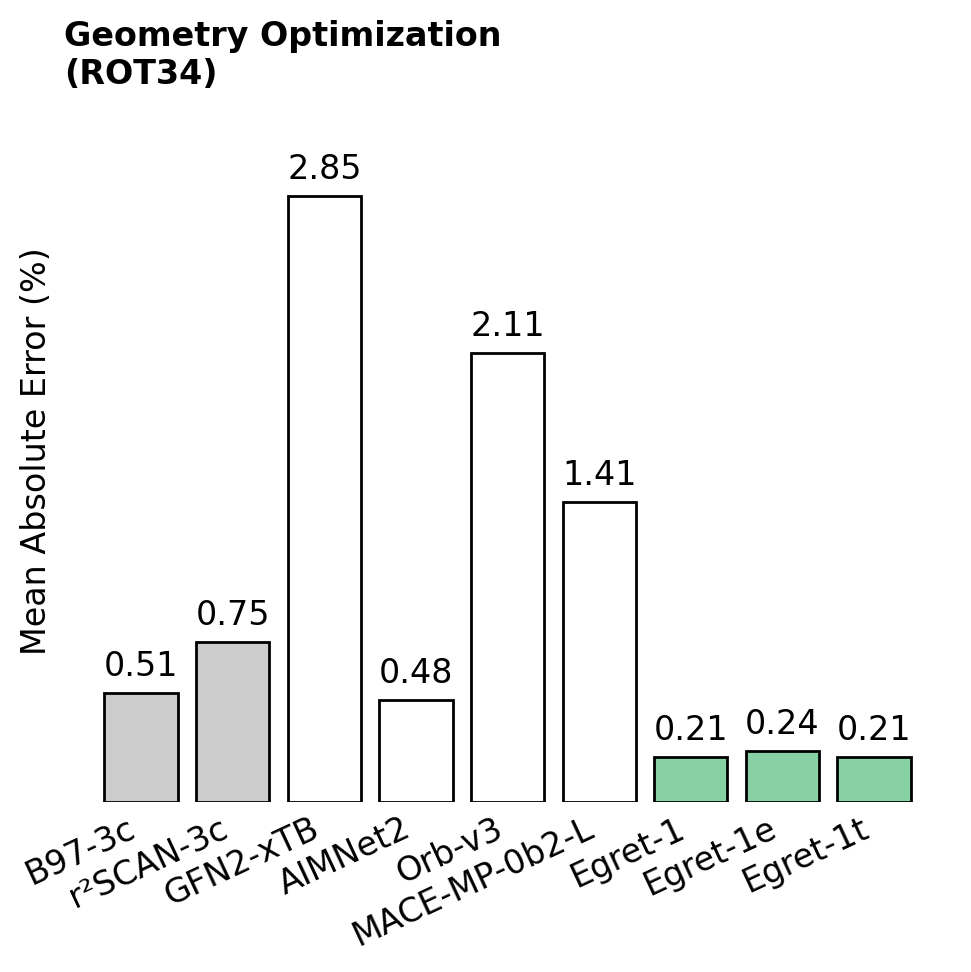
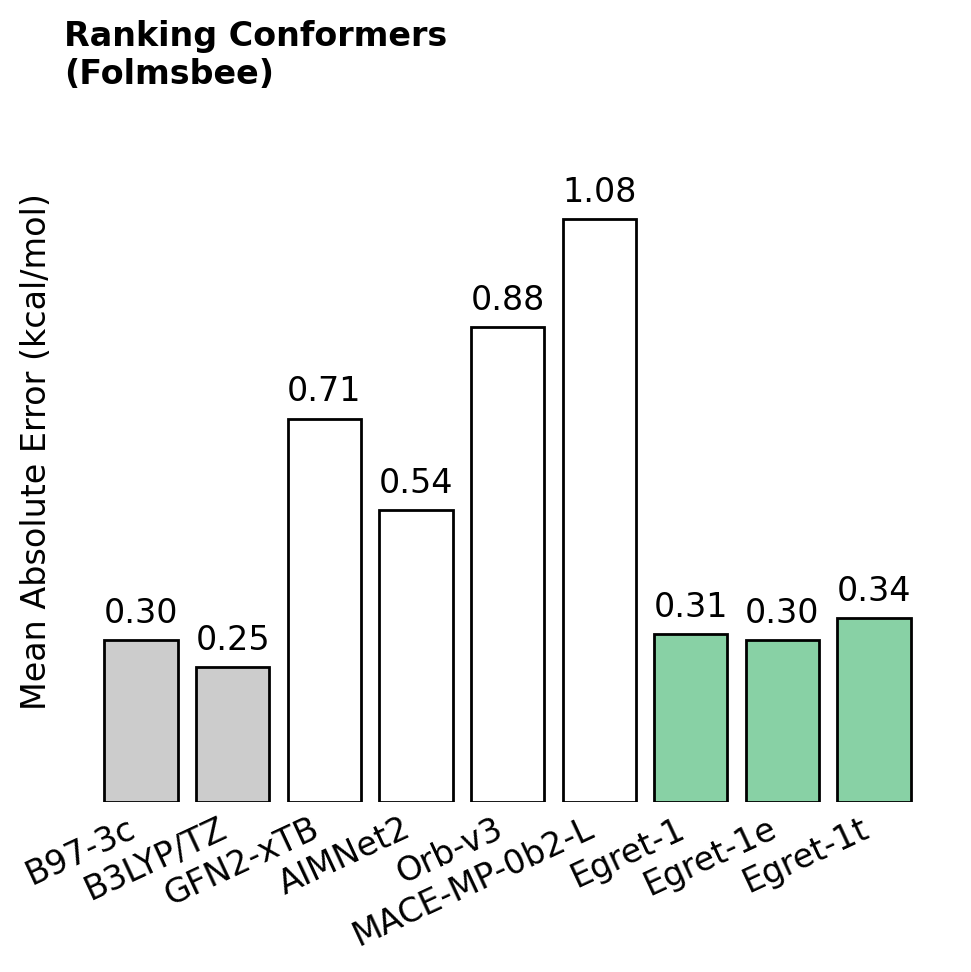
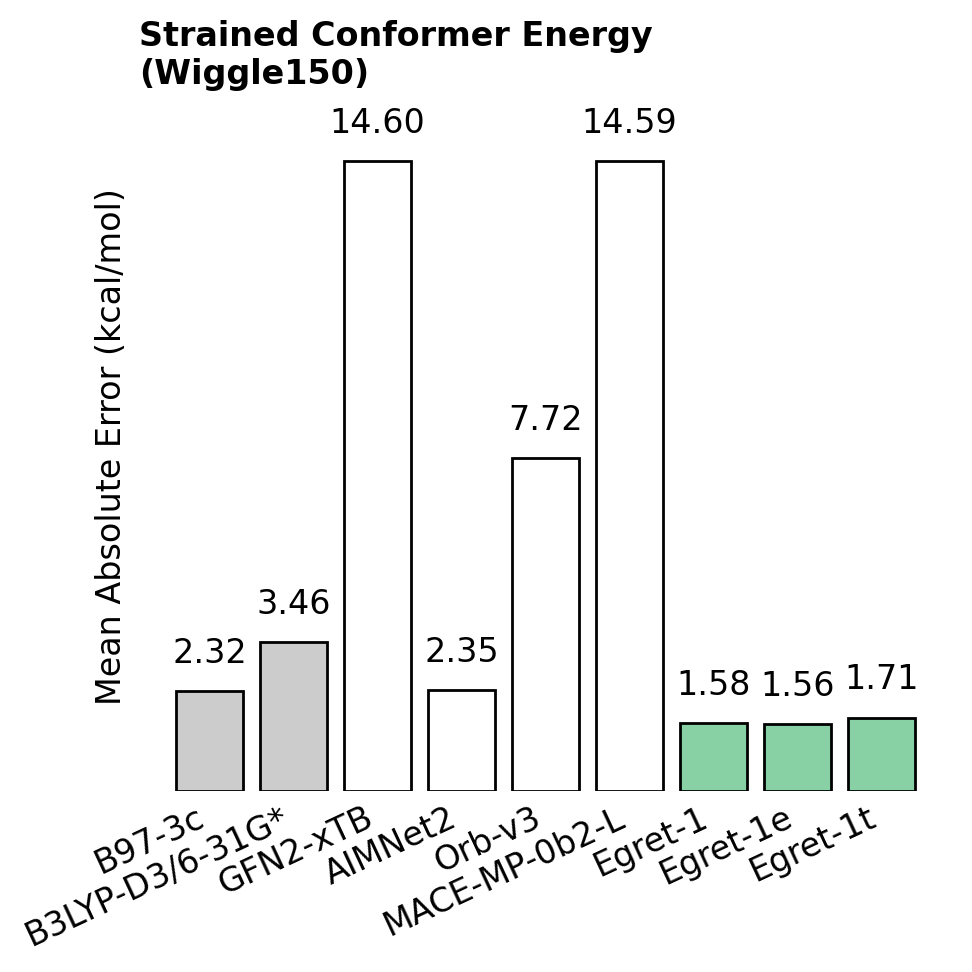
We've benchmarked the Egret-1 models on a wide variety of common simulation tasks, and found that in many cases they exceed the accuracy of conventional quantum-chemical methods—meaning that Egret-1 is both faster and more accurate than the previous state-of-the-art. (For a full description of our training and benchmarking work, we've published a preprint on arXiv.)
Here's three particularly exciting results—in all cases, lower numbers are better.
Chemical accuracy hundreds of times faster
Even on CPUs, the Egret-1 models are far faster than quantum chemistry. A single energy calculation on the macrocyclic drug rapamycin takes almost 15 minutes with a low-cost quantum-chemical method, while the same calculation takes less than two seconds with Egret-1:
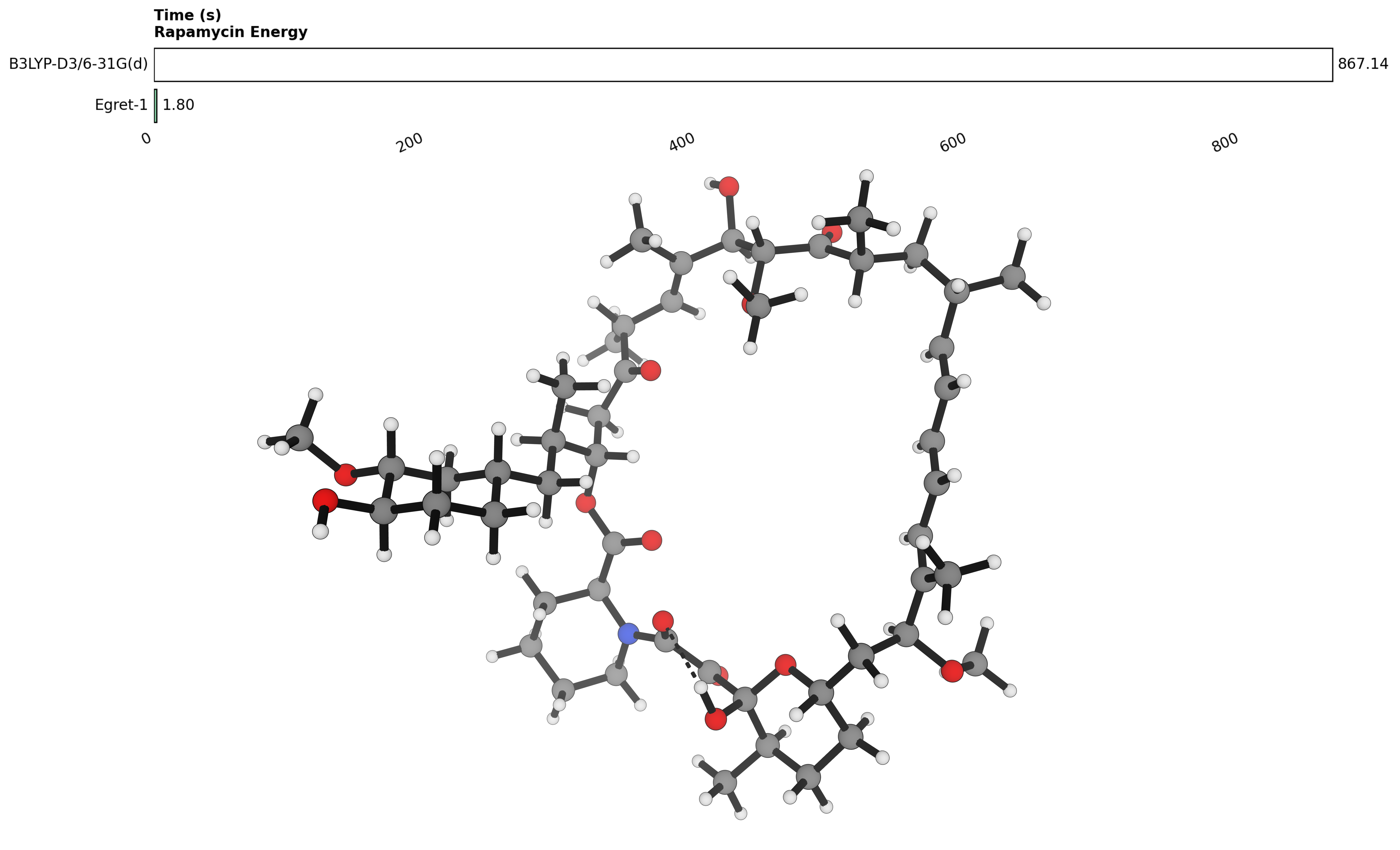
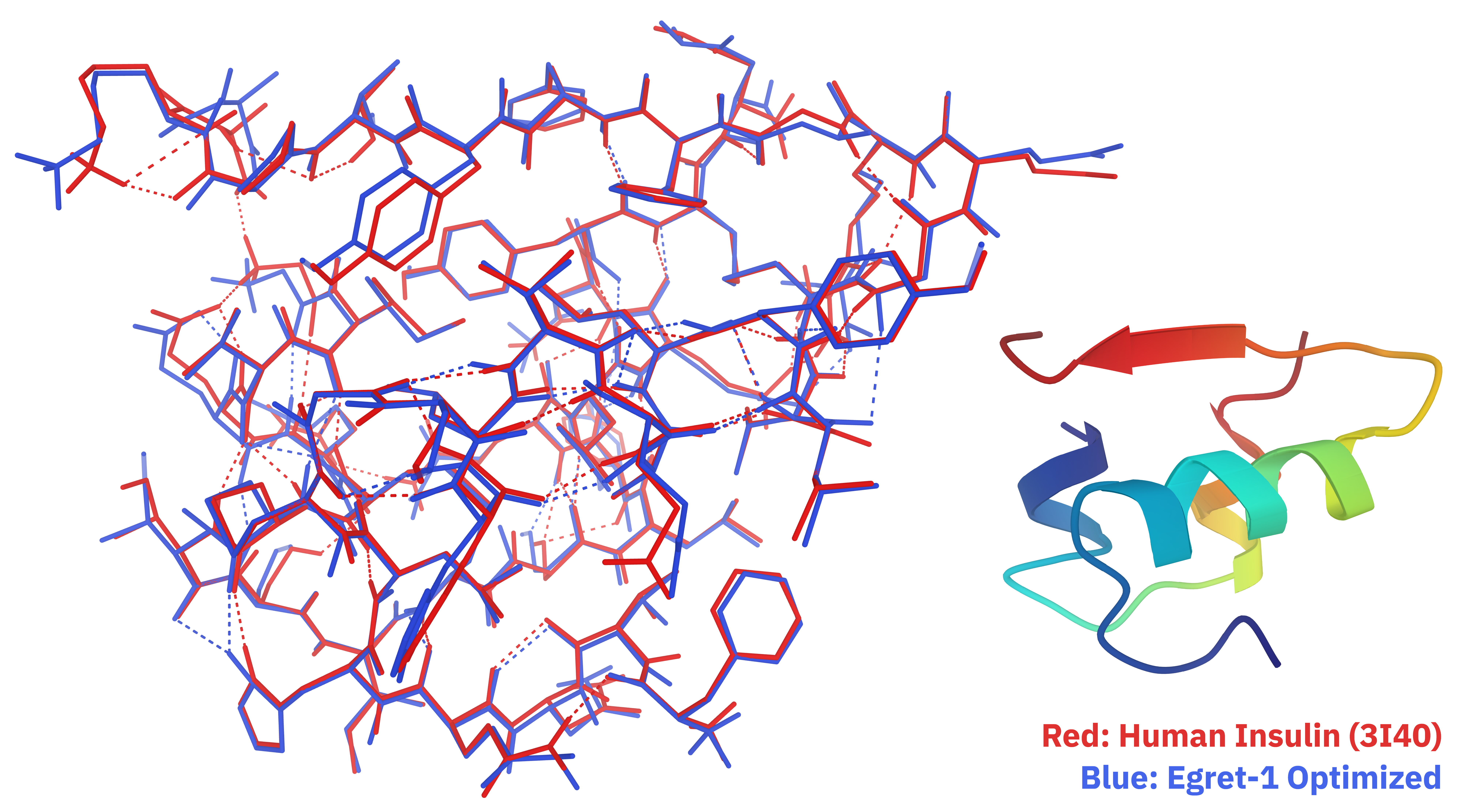
For further speed accelerations, the Egret-1 models can also be run on GPUs. On an H100, Egret-1 can optimize an all-atom structure of insulin in less than three minutes:
How to use Egret-1
We're releasing the Egret-1 models under an open-source MIT license to make it easy to build atop our work. The compiled models can be downloaded from GitHub, and are compatible with the Atomic Simulation Environment. If you're developing with the Egret-1 models, be sure to join the Rowan Discord server to connect with our team.
You can also use Egret-1 through the Rowan computational-chemistry platform. The Egret-1 models can be used for conformer searches, 1D- and 2D-scans, transition states, and more. Make a Rowan account to start running Egret-1 calculations for free today!
What's next
These models have some limitations. Egret-1 is limited to simulating neutral closed-shell structures, supports only a subset of the periodic table, fails to fully learn complex non-covalent interactions, and cannot yet account for solvent effects. We also expect that real-world testing of Egret-1 will find lots of strange and incorrect behaviors we haven't found yet. We plan to address all these limitations with future generations of models, and continue training, scaling and improving general-purpose models for accurate simulation of molecules and materials.










