Representing Local Protein Environments With Atomistic Foundation Models
by Meital Bojan and Sanketh Vedula · Jun 20, 2025
This is a guest post from the authors of a recent arXiv paper that uses embeddings from Egret-1 and other NNPs to study protein environments. Enjoy!
Understanding proteins begins with understanding their smallest parts. Imagine zooming into a protein with a molecular camera, focusing on just a few atoms and the space around them. That tiny neighborhood—defined by nearby atoms, bonds, and interactions—often holds the key to what the entire protein does. Whether it is folding correctly, binding a ligand, or carrying out a reaction, much of a protein's function is shaped locally. If we can accurately capture and represent these environments, we unlock powerful new tools: predicting chemical shifts from structure, identifying functional sites, comparing similar motifs, and even designing new proteins from scratch. But describing these regions computationally is no small feat—they're diverse, dynamic, and chemically complex.
In our recent work, we introduce a new way to represent them: using the internal embeddings of pretrained atomistic foundation models (AFMs)—also known as neural network potentials (NNPs)—such as Egret, Orb, etc. These models are trained to approximate some complicated quantum mechanical property (usually, the ground state energy obtained from numerical approximations to the Schrödinger equation). The key motivation for their development by the computational chemistry community is for quantum-mechanically accurate atomistic simulations. Having a neural proxy for the energy function can allow them to compute derivatives with respect to the atom positions, and it is many orders of magnitude faster than performing DFT-based simulations.
However, our key idea in this paper is this: while trying to approximate this ground-state energy over many millions of atomistic configurations, these AFMs learn rich feature representations. In particular, we noticed that these representations act like high-resolution snapshots, encoding both local geometry and chemistry in a compact, reusable form. In the sections below, we highlight how this opens up new possibilities—from decoding local structure to making highly accurate predictions—all through the lens of AFM embeddings.
Each local protein environment is centered on a residue and includes all nearby atoms. An atomistic foundation model (AFM) embeds this environment, producing a set of rich descriptors for key atoms. These serve as compact, reusable inputs for downstream models that predict structural or chemical properties.
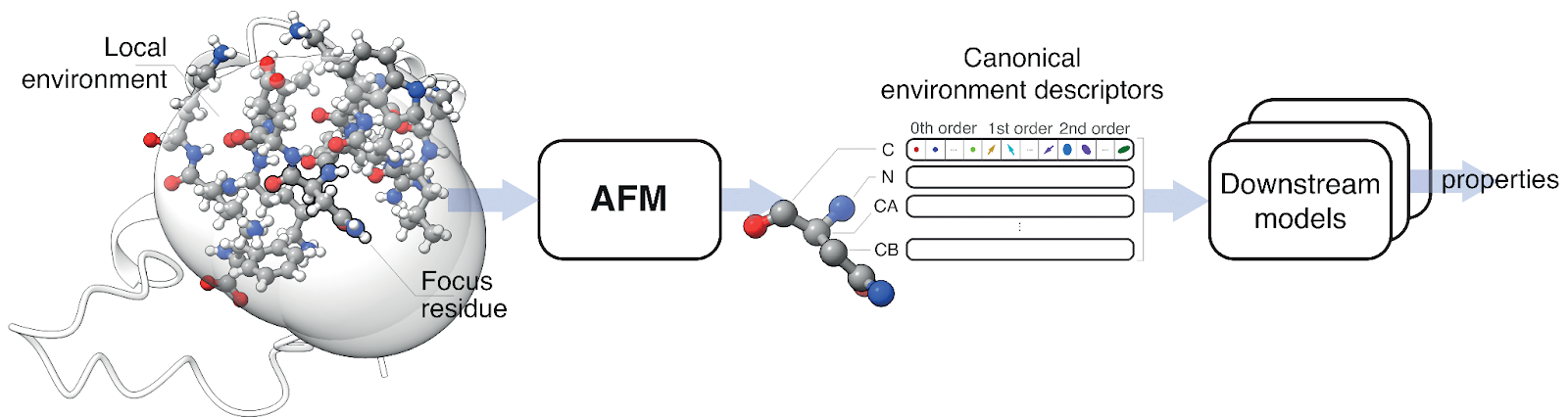
Each local protein environment is centered on a residue and includes all nearby atoms. An atomistic foundation model (AFM) embeds this environment, producing a set of rich descriptors for key atoms. These serve as compact, reusable inputs for downstream models that predict structural or chemical properties.
One Embedding to Rule Them All: Structure and Chemistry
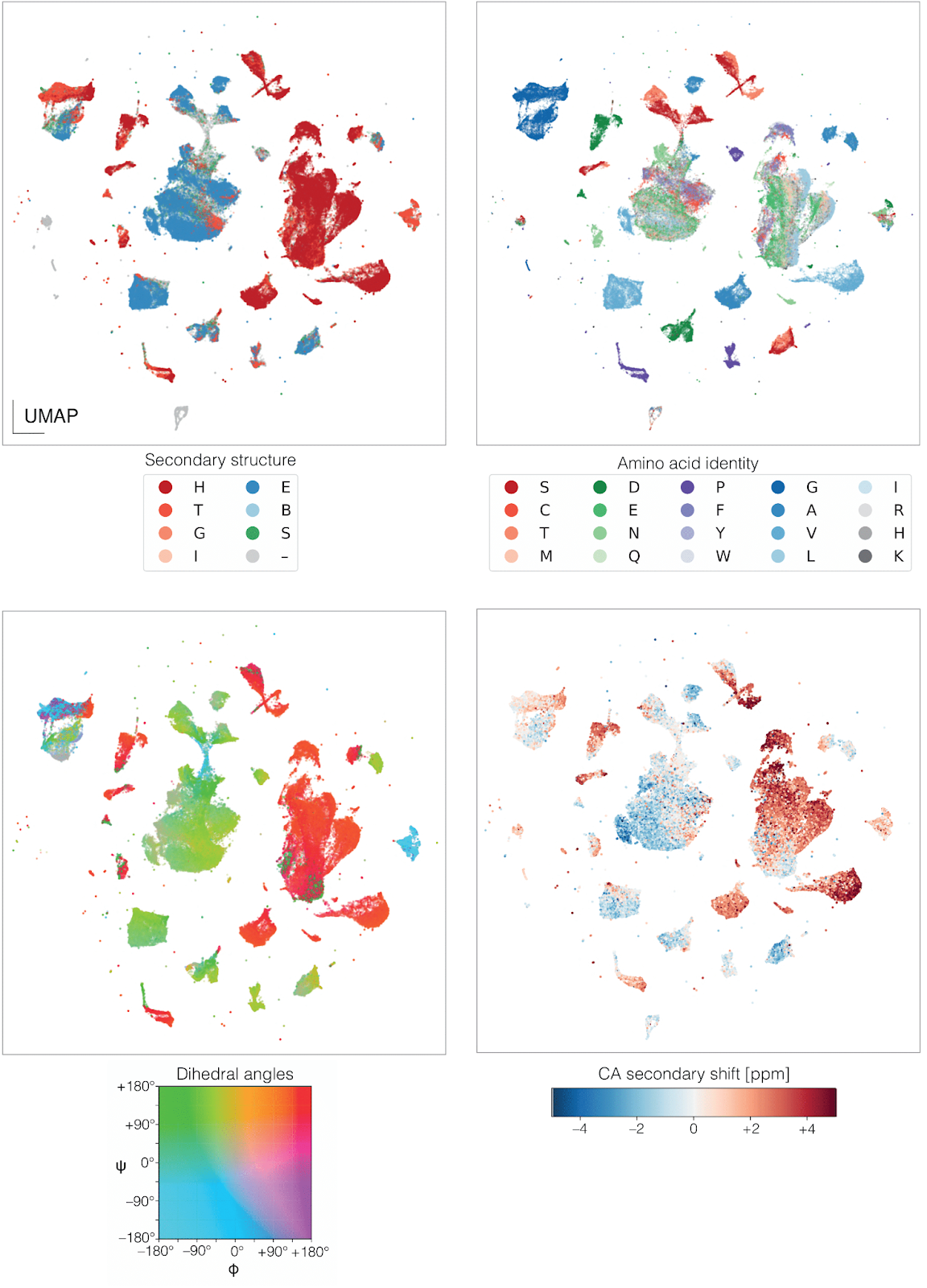
We found that these embeddings naturally reflect both geometric and chemical properties of protein environments. In the plot below, you can see a UMAP projection of the embedding space—colored by secondary structure, amino acid identity, dihedral angles, and even secondary chemical shifts. These patterns emerge without any task-specific supervision, suggesting that AFMs are already encoding the key features we care about. And it doesn't stop there: in the full paper, we show the same embeddings can also predict protonation states—another chemically nuanced property—better than specialized models.
What's Similar, What's Likely, What's Physical
Given a protein structure, how do you know if it looks natural? It turns out the embedding space learned by AFMs is structured enough to answer that. You can measure how similar two environments are—or how likely one is, given what's typical. This opens doors to detecting unusual regions in proteins, assessing structure quality, or even generating new environments that follow the rules of biophysics.
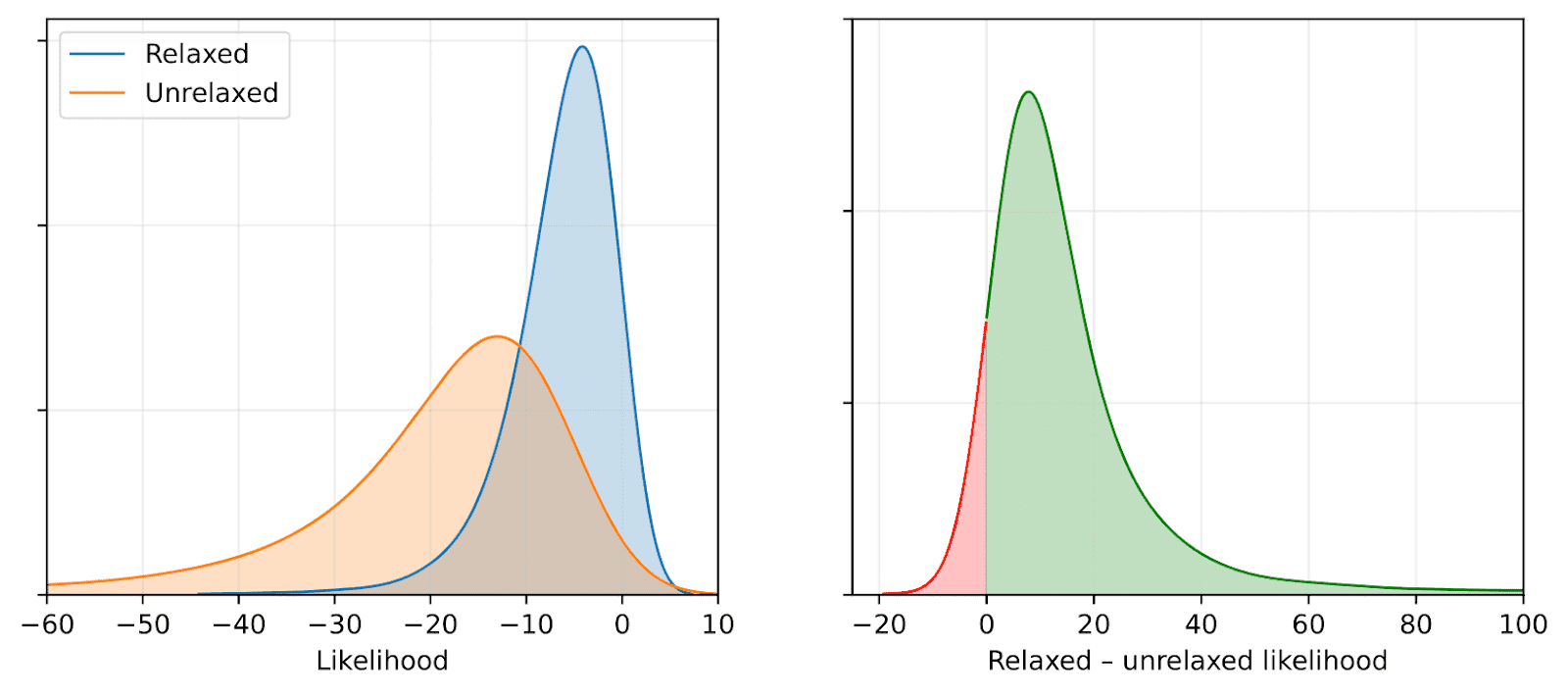
A small structural relaxation shifts the likelihoods of local environments—showing that the model picks up on subtle, meaningful changes in protein geometry.
Next-Level Chemical Shift Prediction
The relation between structure and chemical shifts is notoriously complex due to its inherently quantum nature and accurate prediction of these quantities is an important methodological bottleneck in protein NMR spectroscopy. Using AFM embeddings as inputs, we trained a lightweight model (0.49M parameters trained over 80000 samples) to predict NMR chemical shifts—and it outperforms current state-of-the-art tools. Even better, it captures subtle physical effects, like how aromatic rings alter nearby signals, with a realism that hand-crafted methods struggle to match. On top of that, our model doesn't just make predictions—it also knows when to be uncertain. By leveraging the AFM embedding space, we estimate a confidence score for each predicted shift, giving users a built-in way to assess reliability.
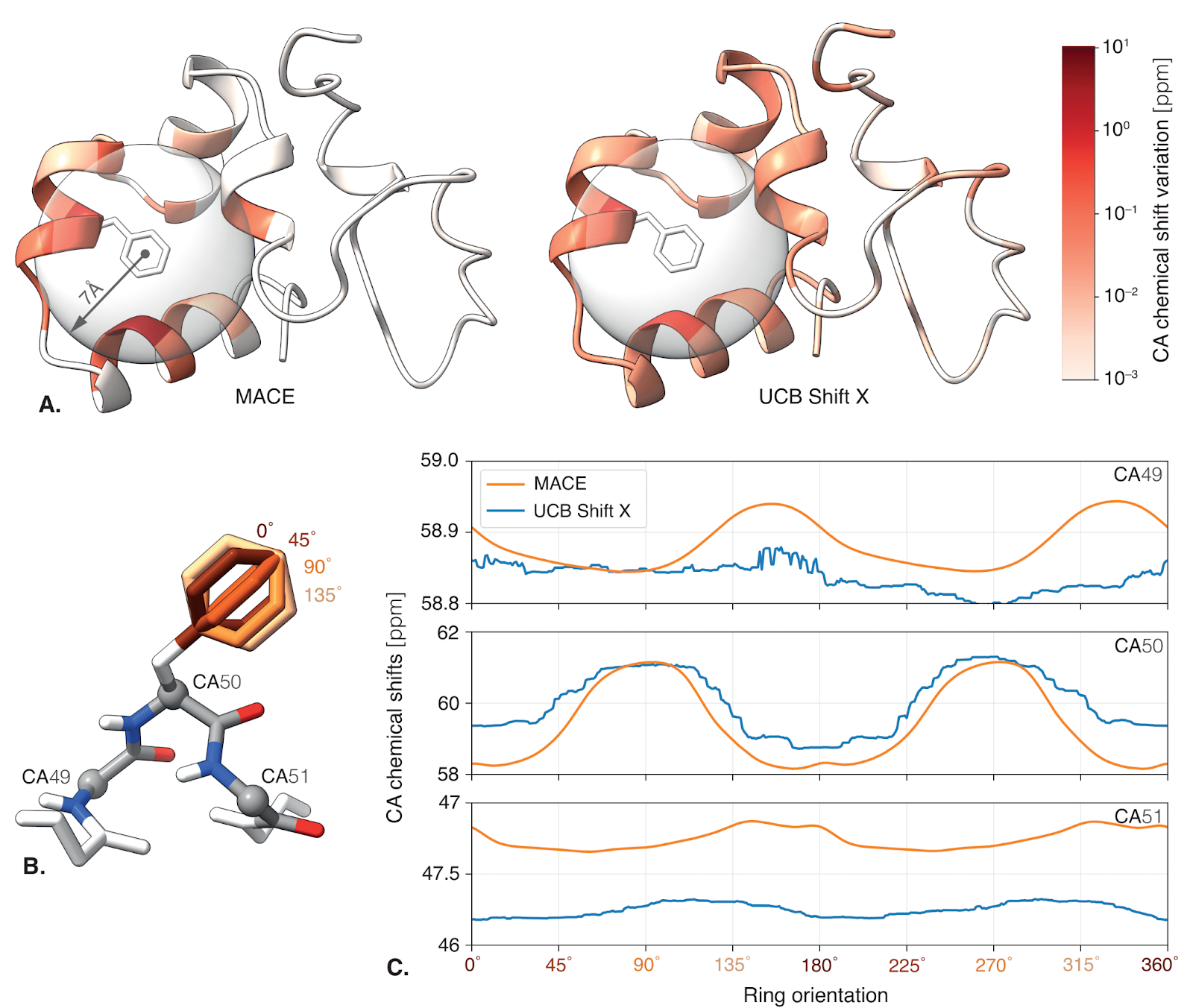
As the aromatic ring rotates, nearby chemical shifts vary in a smooth, physically realistic way—just as expected from ring current effects. The MACE-based predictor captures this behavior with clear 180° periodicity and distance-dependent decay, while traditional methods show less structured or overly long-range influence.
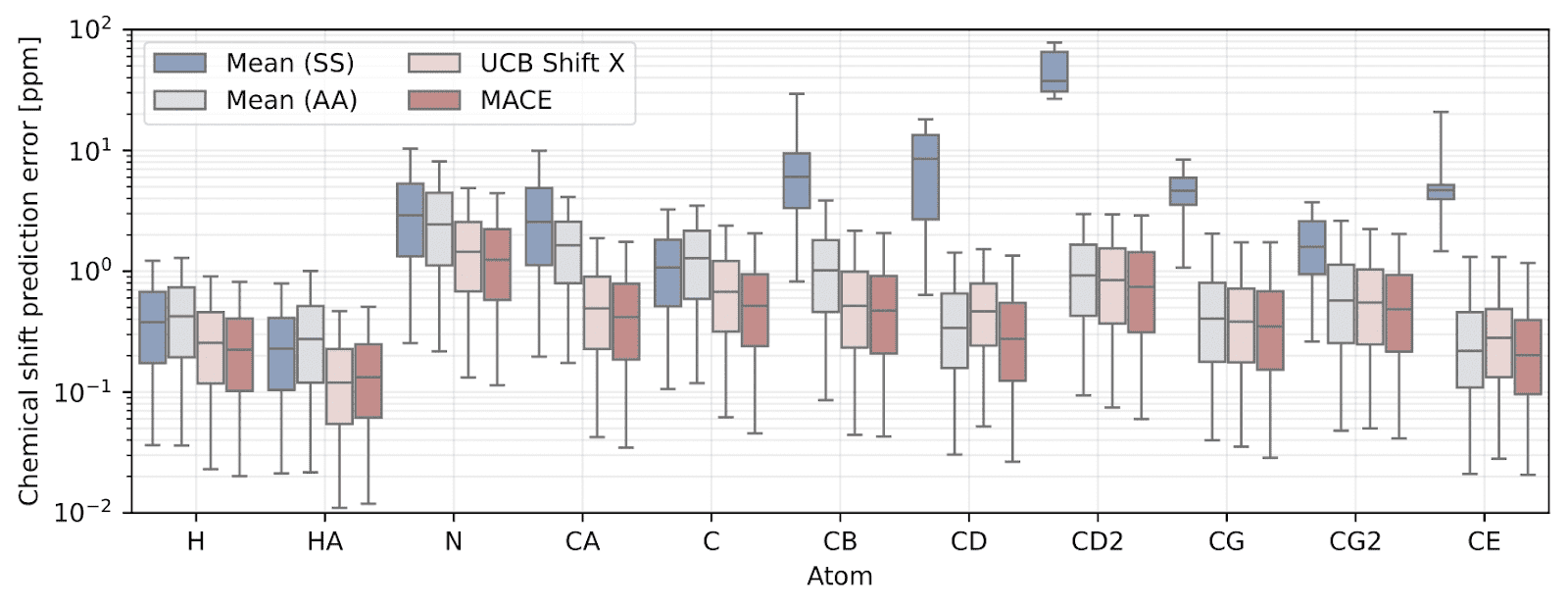
On a large benchmark of real protein environments, the MACE-based predictor consistently outperforms the current state-of-the-art (UCB Shift X) across nearly all atom types—delivering lower prediction errors and tighter confidence intervals.
Peeking Into the Black Box
What exactly do these embeddings learn? We explored how they respond to small changes, like rotating a side chain or unfolding a helix. By following these shifts in the embedding space, we can start to see how structure maps to representation—hinting at the possibility of decoding these embeddings back into 3D structures. In practice, this is done by computing embeddings for both the AlphaFold3 (AF3) prediction and the target structure, then optimizing the atomic positions of the AF3 structure so that its embedding matches the target. Since the embeddings are derived from atomic arrangements, this reverse mapping would effectively mean recovering local structure from the information they compress—a kind of structural inversion that could open up new ways to analyze or even generate protein conformations.
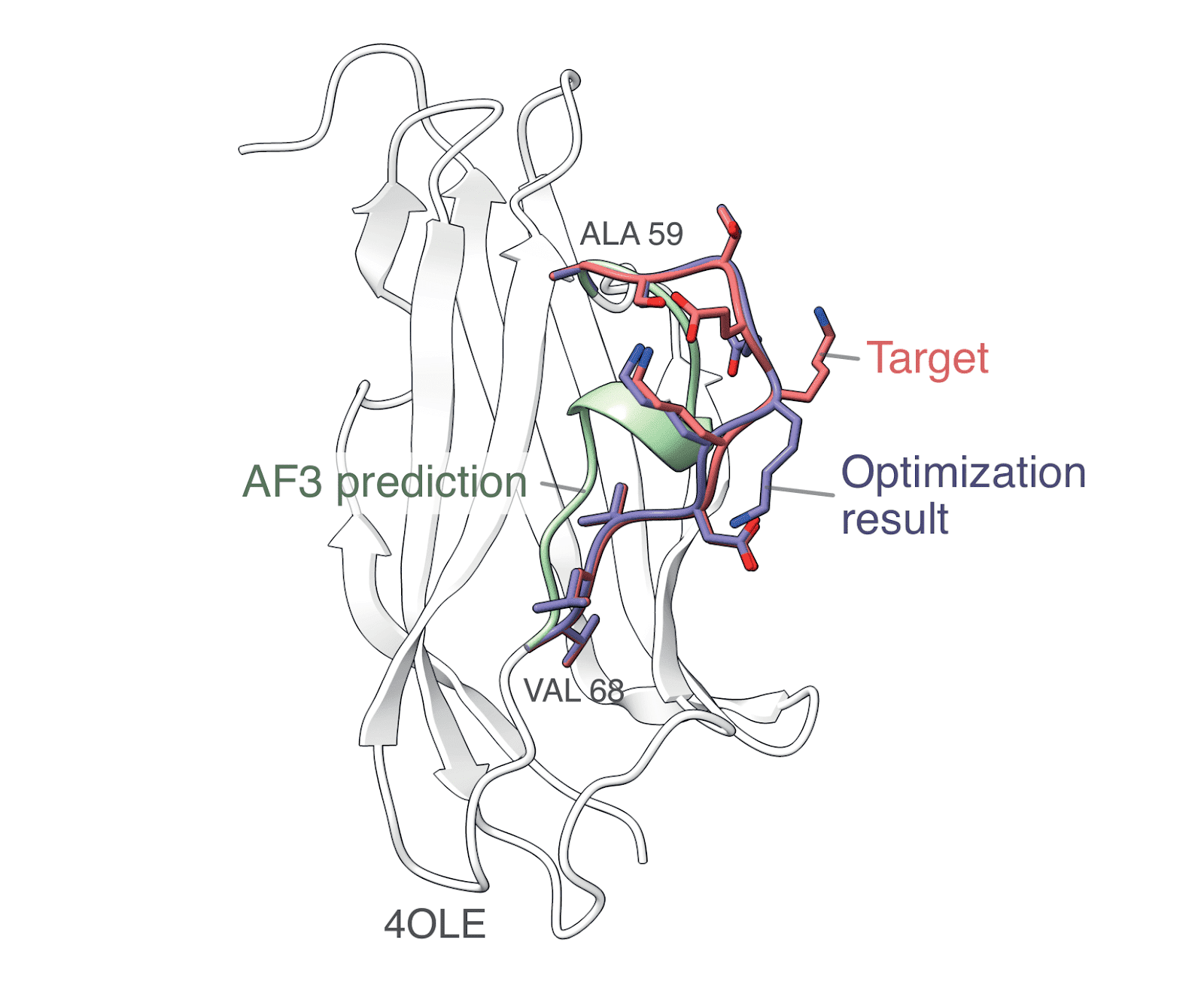
MACE descriptors are used to refine an AlphaFold3 prediction (green), producing a structure (blue) that shifts toward the experimental conformation (red). While not exact, the result shows the potential of these embeddings to be decoded back into meaningful structural detail.
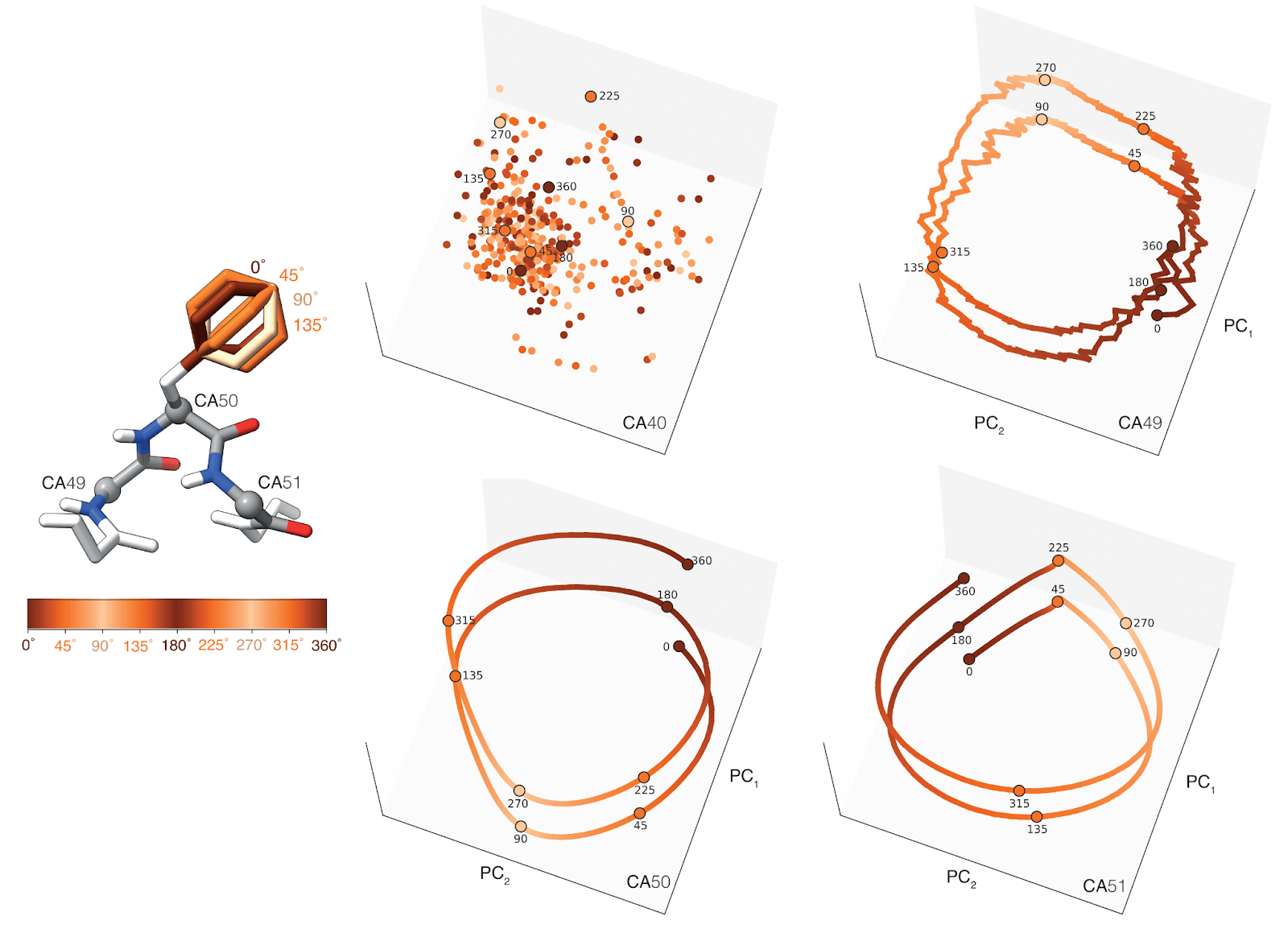
We rotated an aromatic side chain and tracked how the embeddings of nearby atoms changed. The result: smooth, structured paths in the latent space that mirror the physical rotation—like watching the model "feel" the twist. Each point here is a PCA projection of an atom's embedding as the ring rotates, showing just how sensitive and structured these learned representations really are.
Putting AFMs to the Test
We didn't just use one model—we tested several. Among Egret, MACE, OrbNet, and AIMNet, we found that Egret embeddings perform best overall, though AIMNet shows particular strength in protonation prediction. These results hint at different strengths for different AFMs depending on the application.
Together, these findings demonstrate that AFM embeddings aren't just compact descriptors—they're powerful, information-rich representations that capture the shape, chemistry, and subtle variations of local protein environments. Still, this is only the beginning. Although the current outcomes are encouraging, there remains significant potential for further advancement. Our models use frozen embeddings from atomistic foundation models—but fine-tuning them for specific tasks could push performance even further. And while our chemical shift predictor sets a new standard across most atoms, it still trails behind on a few, like HA—highlighting areas for improvement. Looking ahead, the differentiable nature of our predictor opens up exciting possibilities, like guiding structure refinement or helping models like AlphaFold learn directly from experimental NMR data. If you're curious about the technical details, benchmarks, or what's next, check out the full paper.










