Rowan Research Spotlight: Turki Alturaifi
by Corin Wagen · May 7, 2025
What makes a reaction selective? Can we predict, before doing an experiment, where a catalyst will bind or whether a mutation will improve enzyme activity?
These are the kinds of questions that drive the research of Turki Alturaifi, a fourth-year PhD candidate at the University of Pittsburgh in Professor Peng Liu's group. Originally from Saudi Arabia, Turki was first introduced to computational chemistry as an undergraduate working with Rob Paton at Colorado State University. "There was a seminar by Rob about computational chemistry… that was the first time I heard about it," he said. "What caught my eye was how much money and time you could save—and the chemical waste." From that moment on, he was hooked.

Turki Alturaifi
Reaction Mechanisms, Enzyme Catalysis, and Everything In Between
Turki's current research spans both organic and enzymatic catalysis, unified by a central theme: predicting reactivity and selectivity from first principles. "The general overview," he explains, "is how can we predict reactivities and selectivities in organic reactions?" That often means elucidating reaction mechanisms—especially for challenging systems like alkene functionalizations—and using that insight to design new directing groups, new reactions, or better catalysts.
Some of the most complex systems he's studied are enzymes. "There are a lot of libraries that generate combinatorial mutations," he explains, "and one project I'm working on, in collaboration with Professor Yang Yang, is how can we incorporate quantum chemistry information to give you better scoring." The vision is compelling: combining short molecular dynamics simulations with high-level quantum-chemical calculations to predict how protein dynamics might impact catalytic activity. "We're trying to see if the loop flexibility has any relation to the observed enantioselectivity," he says. "It's very difficult… it's really not straightforward to design these libraries."
Much of the field still relies on sequence-based prediction, but Turki argues that incorporating structure-based insights can make models more useful and more accurate. Starting from AlphaFold models or known structures, his workflow mutates enzymes in silico, runs MD simulations, and attempts to correlate loop behavior with experimental results. It's a computationally demanding approach, but one that reflects the real physical complexity of biocatalysts.
The Heteroaryl Database (HArD): Data at Scale
Turki's also worked on more data-driven projects. "I worked on a heteroaryl database," he said, referring to a dataset of over 30,000 heteroaryl groups for which he and collaborators computed a variety of properties relevant to organic chemistry, like Hammett constants. The goal is simple: "If you're troubleshooting a reaction, how can you quickly obtain these properties?" With this database in hand, researchers can now do just that—query electronic properties of potential substrates and feed that information into predictive models, allowing them to more quickly ascertain substrate trends and build meaningful models of reaction yield and selectivity.
What's Hard About Computational Chemistry?
When asked what makes computational chemistry uniquely difficult, Turki doesn't hesitate. "Accuracy," he says. "[A computational prediction] is not an experimental value. It's a computed value. Is it accurate or not? That alone can be a whole significant work," he explains.
Even if Turki can eventually find a way to obtain accurate predictions, he also wants to gain mechanistic insights. "We always try to know the 'why,' and that's much harder," he says. Mechanistic inference is a nuanced process, and Turki often has to evaluate five to ten possibilities before he can propose a single pathway.
Considerations like conformational flexibility can make these studies even harder. "Catalysts are getting bigger. It's very difficult," he says. "What looks easy for 50 atoms becomes impossible for 250." In one recent project studying rhodium catalysts (in collaboration with Professors Jamal Musaev and Huw Davies), Turki turned to QM/MM with xTB and molecular dynamics to understand how π-stacking interactions governed selectivity. Initially, the calculations weren't fast—"27 days to run 500 ps," he admits—but they provided valuable insight into the spatial dynamics of these large, flexible systems.
The Role of Rowan
Turki is a fan of how Rowan makes it easy to employ state-of-the-art methods, days or weeks after they're originally published. "I love the innovations," he says, even though he and his group have their own resources and don't primarily use Rowan for their day-to-day research.
He's used Rowan's ML-based pKa model in a paper currently under review, a collaboration with Professor Keary Engle at Scripps. "We wanted to know if this sulfonyl fluoride is acidic enough to be deprotonated," he said. With Rowan's pKa model "you can test it in two seconds, it's perfect." The result helped explain a surprising selectivity in an oxidative-addition reaction, which reviewers questioned during peer review. "The reaction works with sulfonyl fluorides, but not with iodides. We knew from experiment, but this confirmed it."
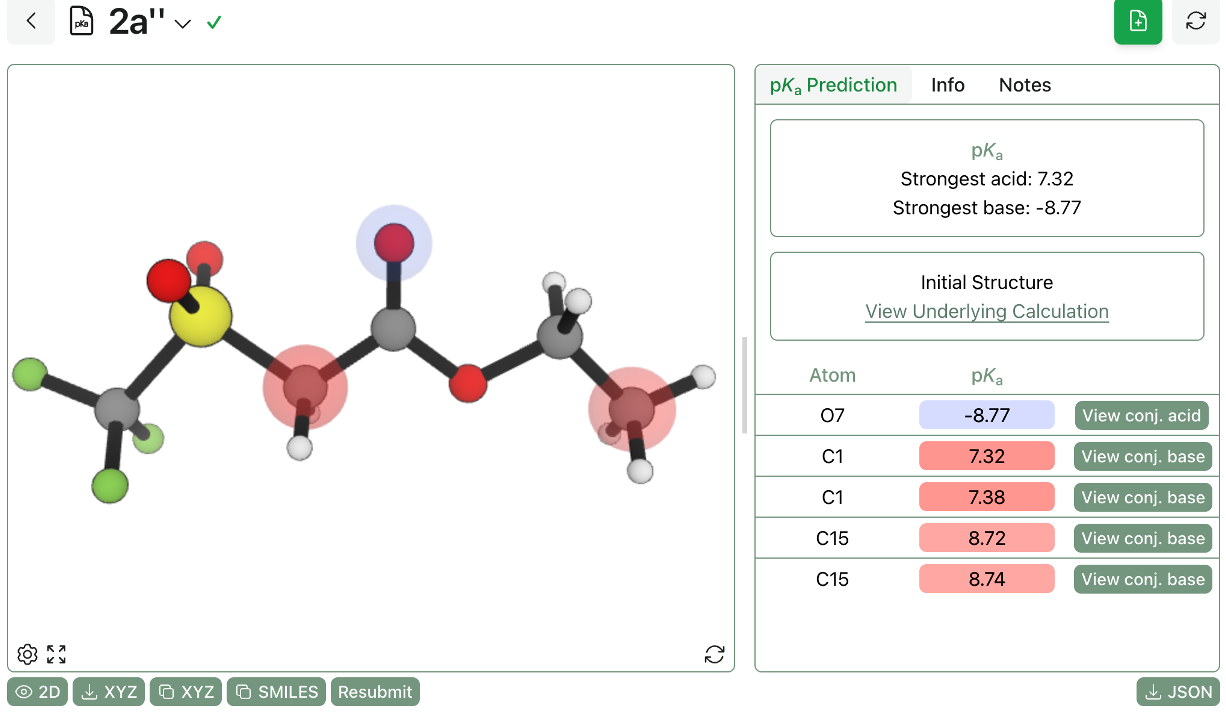
Turki's pKa calculations help to explain surprising selectivity in oxidative addition.
Turki appreciates that Rowan emphasizes transparency and reproducibility: "Rowan gives you optimized workflows you can trust. There are citations, references—it's not like it's pulled from nowhere." And while he's comfortable working in a terminal, he knows that's not true for every chemist. "The difficulty is not opening the terminal—it's the time. As an organic chemist, should you do B3LYP or ωB97X-D? Do you have time to look into this? Rowan gives you the default."
Looking Ahead: A Dream of De Novo Reaction Design
What excites Turki about the future? "The equivalent of de novo protein design for organic molecules," he says. "That's a big dream I have." He hopes for a world where researchers can put in a reaction and receive a predicted substrate scope—or better yet, a predicted catalyst.
But he's skeptical of one-size-fits-all automation. "We don't want to do the decision-making or inference for the scientist," he says. Instead, the goal should be to empower researchers with better tools. "Providing the tools is everything," he says, highlighting the value of software like Rowan.
In the meantime, Turki is keeping busy. He's preparing a revision for Nature Synthesis, working on a follow-up to the heteroaryl database, and building models to predict enzyme activity from structure and quantum descriptors. As the tools improve, and as platforms like Rowan lower the barrier to computational chemistry, Turki's vision of data-rich and mechanism-guided catalyst design gets closer to reality.










