Exploring Meta's Open Molecules 2025 (OMol25) & Universal Models for Atoms (UMA)
by Corin Wagen and Ari Wagen · May 23, 2025
Last week, Meta's Fundamental AI Research (FAIR) team released Open Molecules 2025 (OMol25), a massive dataset of high-accuracy computational chemistry calculations. In addition to the dataset, they released several pre-trained neural network potentials (NNPs) trained on the dataset for use in molecular modeling, including a new "Universal Model for Atoms" (UMA) that unifies OMol25 with other datasets from the FAIR-chem team.
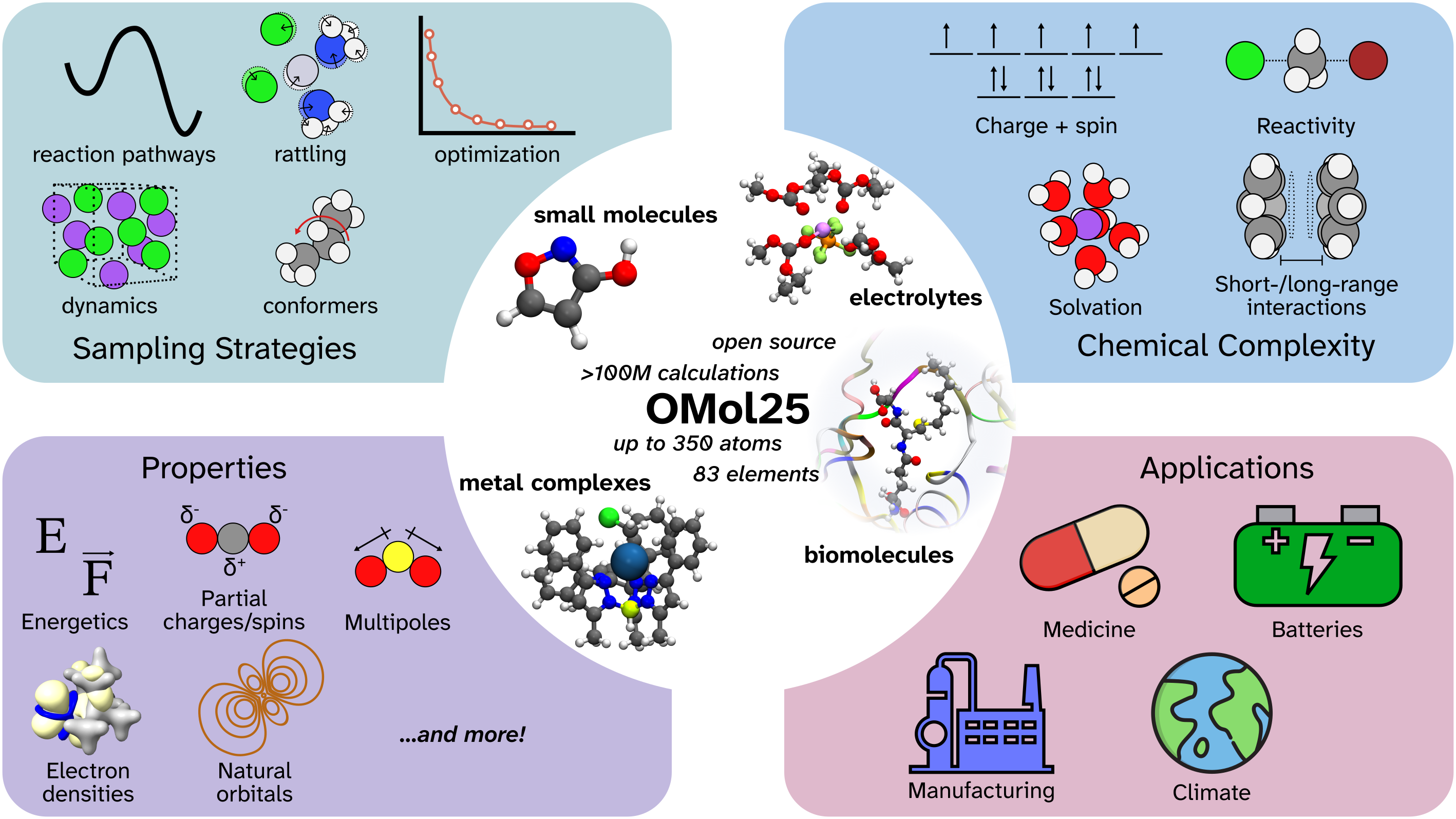
A visual overview of OMol25. Image taken from Figure 1 of Levine et al. 2025.
We've been following this space pretty closely for a while, and I (Corin) had the privilege of being one of the few non-Meta chemists in the room when these models were announced at the Meta Open-Source AI conference in Austin. It was immediately obvious, just from the first few slides, that this would go down as one of the most impactful scientific releases of the year—and that the "ML for atomistic simulation" field would be divided into "pre-OMol25" and "post-OMol25" eras.
This intuition has been confirmed over the last week. Our own internal benchmarks confirm that the models are far better than anything else we've studied, and feedback from scientists indicate that these models are useful in the real world: one Rowan user wrote that the OMol25-trained models give "much better energies than the DFT level of theory I can afford" and "allow for computations on huge systems that I previously never even attempted to compute." Another Rowan user called this "an AlphaFold moment" for the field.
Scientists who've already been working in this field have doubtless already read and digested the papers, which have been omnipresent on social media. But for non-ML-focused scientists, who are just now learning about OMol25 and its implications, it can be confusing to situate these results in the broader scientific context. In this blog post, we'll take a close look at the dataset, the pre-trained models, and some outstanding issues while sharing some thoughts about the future of NNP-accelerated atomistic simulation.
(If you're interested in trying out a model trained on OMol25 right now, you can run simulations using one of Meta's eSEN models on Rowan—make a free account or log in with Google in seconds.)
Background
The OMol25 and UMA releases comprise a (1) dataset of quantum chemical calculations and (2) a number of neural network potentials (NNPs) trained on this dataset. Briefly, the goal of NNPs is to provide a fast and accurate way to compute the potential energy surface of arbitrary molecules or materials, which avoids the shortcomings of both quantum mechanics and forcefield-based approaches.
If you're completely new to this, we suggest you read our introduction to NNPs, which explains the history of the field and the state-of-the-art (as of last year).
Dataset
Previous molecular datasets have been limited by size, diversity, and accuracy. Early datasets like the ANI-1 datasets were run at a relatively low level of theory (ωB97X/6-31G(d)) and contained only simple organic structures with four elements, which limits the applicability of downstream models to lots of important applications.
The OMol25 dataset addresses all these shortcomings. It comprises over 100 million quantum chemical calculations, which in total took over 6 billion CPU-hours to generate, and contains an unprecedented variety of diverse chemical structures. The three areas of particular focus were biomolecules, electrolytes, and metal complexes, but a wide variety of previous community datasets were also compiled to give complete coverage of other areas.
To ensure that the underlying quantum chemistry was sufficiently high-level, all calculations were run at the ωB97M-V level of theory using the def2-TZVPD basis set. ωB97M-V, developed by Narbe Mardirossian and Martin Head-Gordon, is a state-of-the-art range-separated meta-GGA functional, which avoids many of the pathologies associated with previous generations of density functionals (like band-gap collapse or problematic SCF convergence). The calculations were run with a large pruned 99,590 integration grid, which allows for non-covalent interactions and gradients to be computed accurately. (Read our blog post on common DFT errors for why this matters.)
Here's a brief overview of the different areas of chemistry represented in OMol25:
Biomolecules
Biomolecular structures were pulled from the RCSB PDB and BioLiP2 datasets. Random docked poses were generated with smina, and Schrödinger tools were used to extensively sample different protonation states and tautomers. Various environments representing protein–ligand, protein–nucleic acid, and protein–protein interfaces were sampled, and restrained molecular dynamics simulations were run to sample different poses. Non-traditional nucleic acid structures, like triplex systems, Holliday junctions, and A- & Z-DNA structures, were also sampled.
Electrolytes
A variety of electrolytes, including aqueous solutions, organic solutions, ionic liquids, and molten salts, were sampled. Molecular dynamics simulations were run for various disordered systems, including those with novel electrolyte materials, and clusters were extracted (including solvent-only clusters). Various oxidized or reduced clusters relevant for battery chemistry were investigated, as were electrolyte-degradation pathways. Gas–solvent interfaces were also sampled, and ring-polymer molecular dynamics was used to account for nuclear quantum effects.
Metal Complexes
Metal complexes were combinatorially generated using combinations of different metals, ligands, and spin states, with geometries created using GFN2-xTB through the Architector package. Reactive species were generated using the artificial force-induced reaction (AFIR) scheme, which generated a reactive path from which structures could be sampled.
Other Datasets
Existing datasets like SPICE, Transition-1x, ANI-2x, and OrbNet Denali were also recalculated at the same level of theory, which gives the OMol25 dataset solid coverage of main-group and biomolecular chemistry, as well as a substantial number of reactive systems. (Additional reactive structures were generated from the RDG1, PMechDB, and RMechDB datasets to ensure adequate coverage.)
Overall, the OMol25 dataset is 10–100x bigger than previous state-of-the-art molecular datasets like SPICE and the AIMNet2 dataset, and contains much more chemical diversity. This is a tremendous resource for the field, and is likely to drive years of innovations in model development and applications. With the release of OMol25, it's likely that dataset creation will be less important in the short term, and that architectural innovations and applications research will take center stage.
Nevertheless, the scale of the OMol25 dataset will make training challenging for organizations without access to large numbers of GPUs, like academic groups and companies not solely focused on training AI models. Following precedent from other fields, we expect that such organizations will shift their focus to dataset curation strategies, fine-tuning existing models, distillation, and other activities which don't require training massive and expensive models from scratch.
Models
To immediately demonstrate the potential of the new OMol25 dataset, the FAIR Chemistry team and collaborators trained and released a number of models trained on the new data, including several using their recently-developed eSEN architecture and one trained using their new Universal Models for Atoms (UMA) architecture. They also report the performance of a number of pre-trained models that haven't been released yet, including a large eSEN model, medium and large UMA models, a MACE model trained on the neutral parts of OMol25, and a GemNet-OC model.
At a high level, the performance of these models is very impressive. Both the UMA and eSEN models exceed previous state-of-the-art NNP performance and match high-accuracy DFT performance on a number of molecular energy benchmarks that I (Ari) have run.
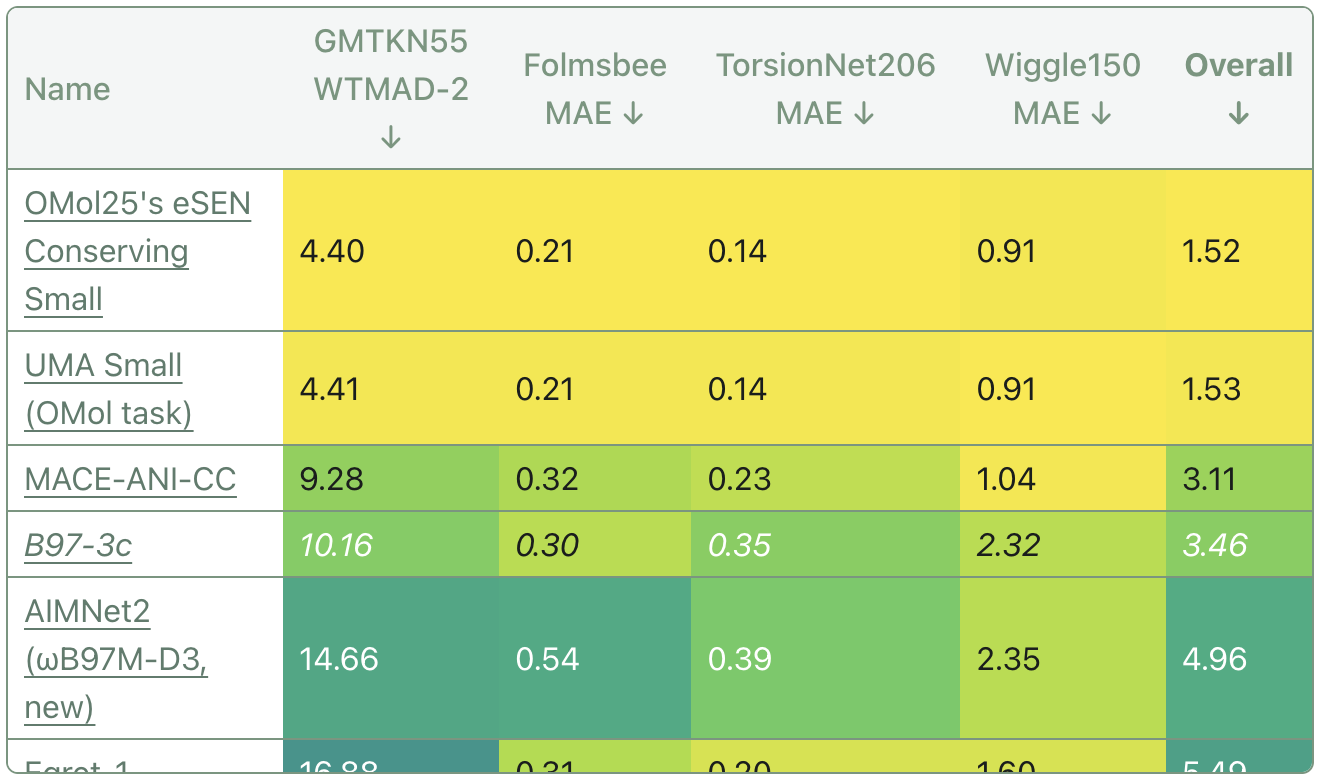
This table is taken from the "Molecular Energy Accuracy" section of Rowan Benchmarks. (The GMTKN55 WTMAD-2 shown here is filtered to only include exclusively neutral, singlet, and elemental-organic subsets.)
The models trained on OMol25 achieve essentially perfect performance on all benchmarks, including on the Wiggle150 benchmark that I (Corin) published just a month ago.
eSEN Direct and Conserving Models
The eSEN architecture (arXiv) was developed by researchers at Meta and released this February. At a high level, eSEN is similar to Equiformer (V1, V2) and eSCN (arXiv), adopting a transformer-style architecture and using equivariant spherical-harmonic representations for some intermediate operations. The eSEN architecture improves the smoothness of the resultant potential-energy surface relative to the previous models, making e.g. molecular dynamics and geometry optimizations better-behaved.
One interesting result from the eSEN paper is that you can speed up conservative-force NNP training by adopting a two-part training scheme:
We start from a direct-force model trained for 60 epochs, remove its direct-force prediction head, and fine-tune using conservative force prediction. The conservative fine-tuned model achieves a lower validation loss after being trained for 40 epochs compared to the from-scratch conservative model being trained for 100 epochs. The fine-tuning strategy also reduces the wallclock time for model training by 40%.
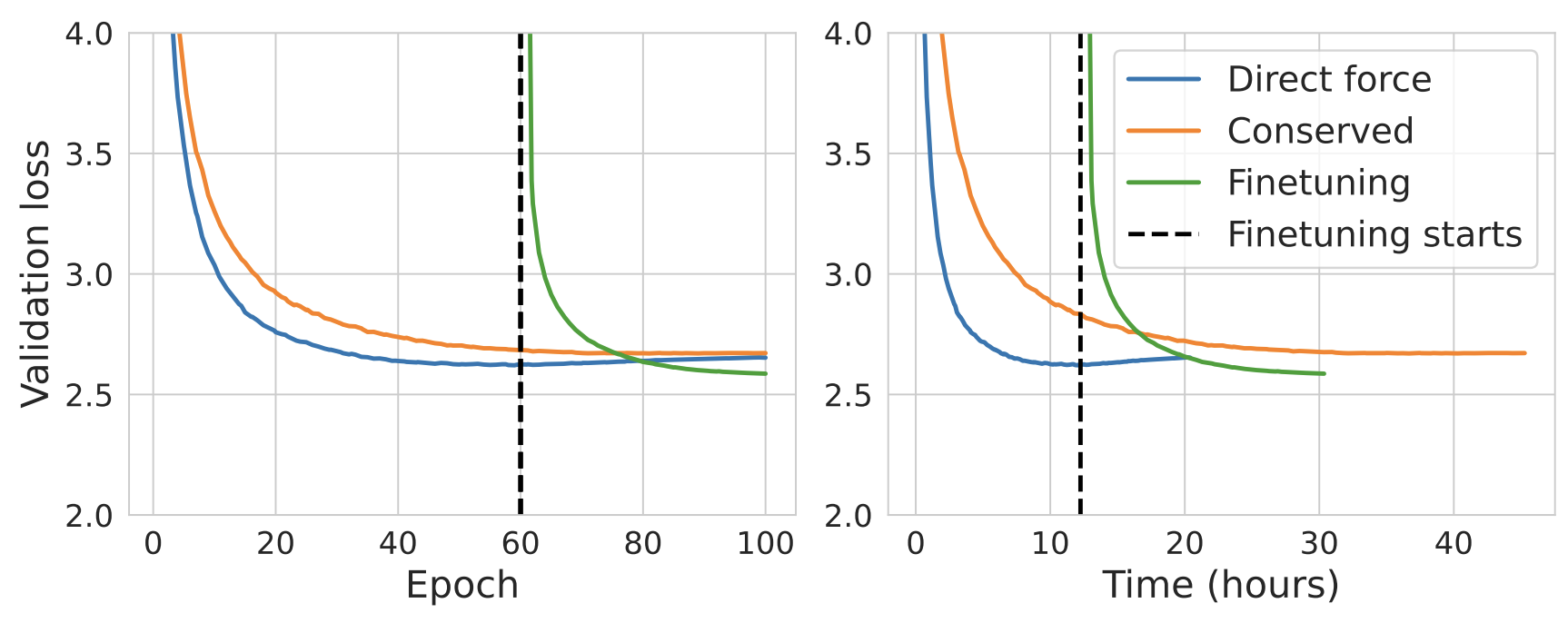
This two-phase training scheme reduces the training times of conservative-force NNPs. Image taken from Figure 3 of Fu et al. 2025.
The OMol25 team trained four models on the new OMol25 dataset using the eSEN architecture: small, medium, and large direct-force prediction models as well as a small conservative-force prediction model. Their findings are conclusively in favor of conservative-force prediction and larger models, though these changes both slow model inference:
Conserving models (cons.) outperform their direct (d.) counterparts across all splits and metrics. Larger models, such as eSEN-md, also outperform their smaller variants.
We chose to list the small, conservative-force prediction eSEN model on Rowan, owing in part to some documented concerns with non-conservative models. All the pre-trained eSEN models (except the large direct-force model, at the time of writing) are available on HuggingFace.
UMA Models
Immediately following the release of OMol25, Meta FAIR announced the release of "UMA: A Family of Universal Models for Atoms." In their preprint, they introduce a new architecture and set of pre-trained models trained on OMol25 as well as the OC20, ODAC23, OMat24, and forthcoming Open Molecular Crystals 2025 (OMC25) datasets.
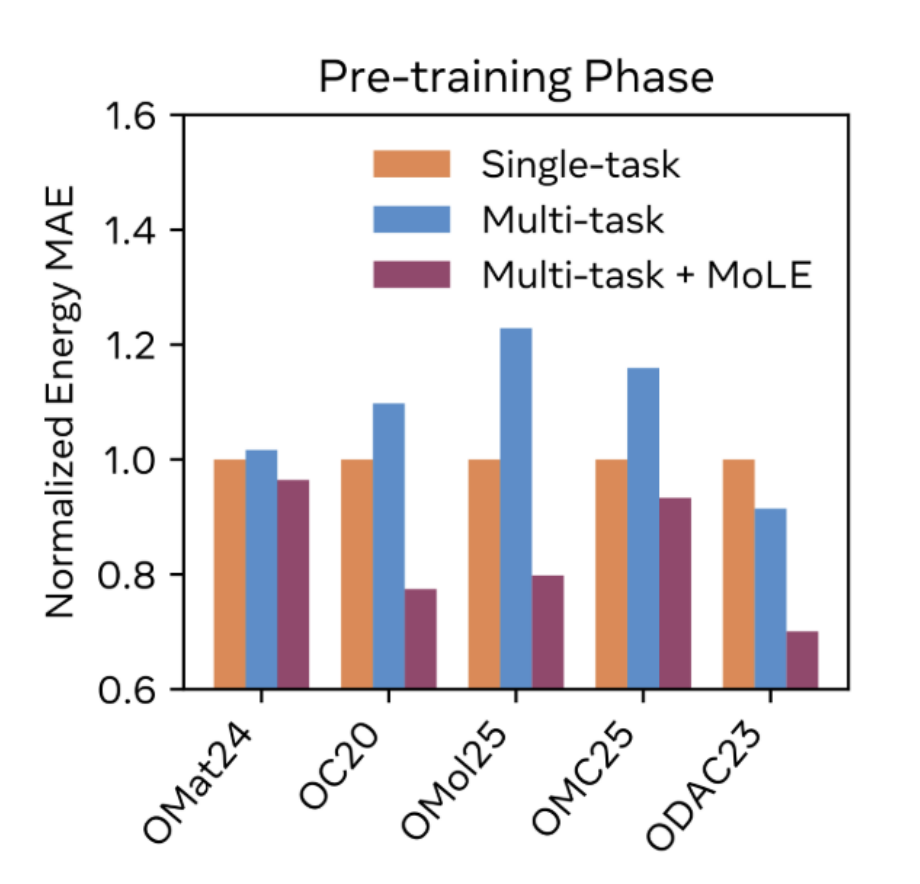
To train UMA models on datasets computed using different DFT engines, basis set schemes, and levels of theory, the authors introduce a novel Mixture of Linear Experts (MoLE) architecture. This MoLE architecture adapts the ideas behind Mixture of Experts (MoE) to the neural network potential space, enabling one model to learn and improve from dissimilar datasets without significantly increasing inference times. The UMA model training also builds on the two-phase training introduced in the eSEN preprint, adding an additional edge-count limitation to the first phase to further accelerate training.
The MoLE scheme dramatically outperforms naïve multi-task learning, and even performs better than a variety of single-task models. This shows that there's knowledge transfer happening across datasets—for instance, adding the other datasets to OMol25 makes the resultant model better at OMol25-related tasks, indicating that the UMA model is learning some general chemical principles.
The UMA models (currently only the small checkpoint) are available on HuggingFace.
GemNet-OC and MACE
The GemNet-OC model reported in the OMol25 paper "outperforms eSEN-sm across all splits, and is comparable to eSEN-md across most metrics." This GemNet-OC model has around the same number of parameters as eSEN-md and a significantly larger cutoff radius (12 Å versus eSEN's 6 Å). A 6 Å–cutoff GemNet-OC model was also trained, which displayed slightly diminished performance.
The MACE model reported in the OMol25 paper seems to underperform eSEN-sm, though the authors don't report many benchmarks.
Shortcomings
The models here don't explicitly incorporate any charge- or spin-related physics, and the authors observe that errors are noticeably higher for systems with net charge or unpaired electrons:
While baseline models have shown strong performance, approaching chemical accuracy (∼ 1 kcal/mol) especially in domains such as biomolecules and neutral organics; OMol25's evaluation tasks reveal significant gaps that need to be addressed. Notably, ionization energies/electron affinity, spin-gap, and long range scaling have errors as high as 200-500 meV.
These results suggest that explicit physics appears necessary even at the large scales of the OMol25 dataset. The present eSEN- and UMA-based models provide a good baseline against which to evaluate future physics-aware schemes; it will be very interesting to see how different approaches to incorporating charge and spin compare on this dataset.
Thoughts
Overall Performance
We've been very happy with using OMol25-based models for routine tasks that we would previously have used DFT for. Here's a system that I (Corin) used to study in the Buchwald Lab at MIT, Pd(II)-mediated C–F reductive elimination:
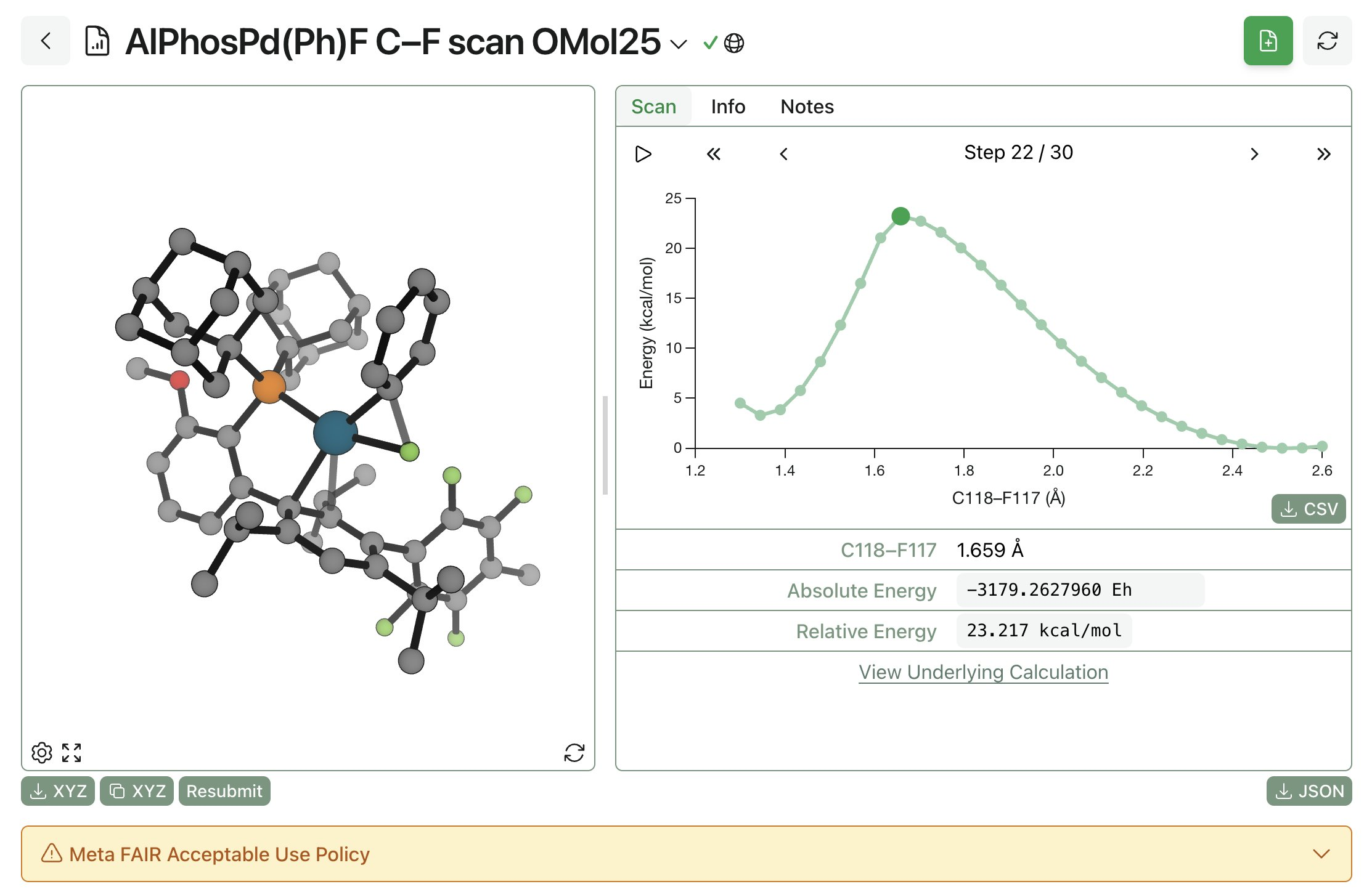
In my tests (documented more fully on X), the Meta model correctly predicted relative barrier heights for C–F reductive elimination, C–O reductive elimination, and different aryl groups. This is a pretty complex system that used to require weeks of CPU time and complex DFT setups; now, this whole scan can be done with a few clicks in an hour or two.
This really is a step change in "how good molecular simulation can be," and I expect that there will be immediate and dramatic effects on research labs across the country.
Towards Multiplicity-Aware Models
The OMol25 dataset provides a bunch of high-accuracy data that we hope will be used to train models that handle spin multiplicity (unpaired electrons) in increasingly sophisticated ways. We look forward to using the eSEN and UMA models as well as any future models trained on OMol25 to accelerate the modeling of radical-containing structures to accurately predict redox potentials, model fragmentation events, calculate bond-dissociation energies, and more.
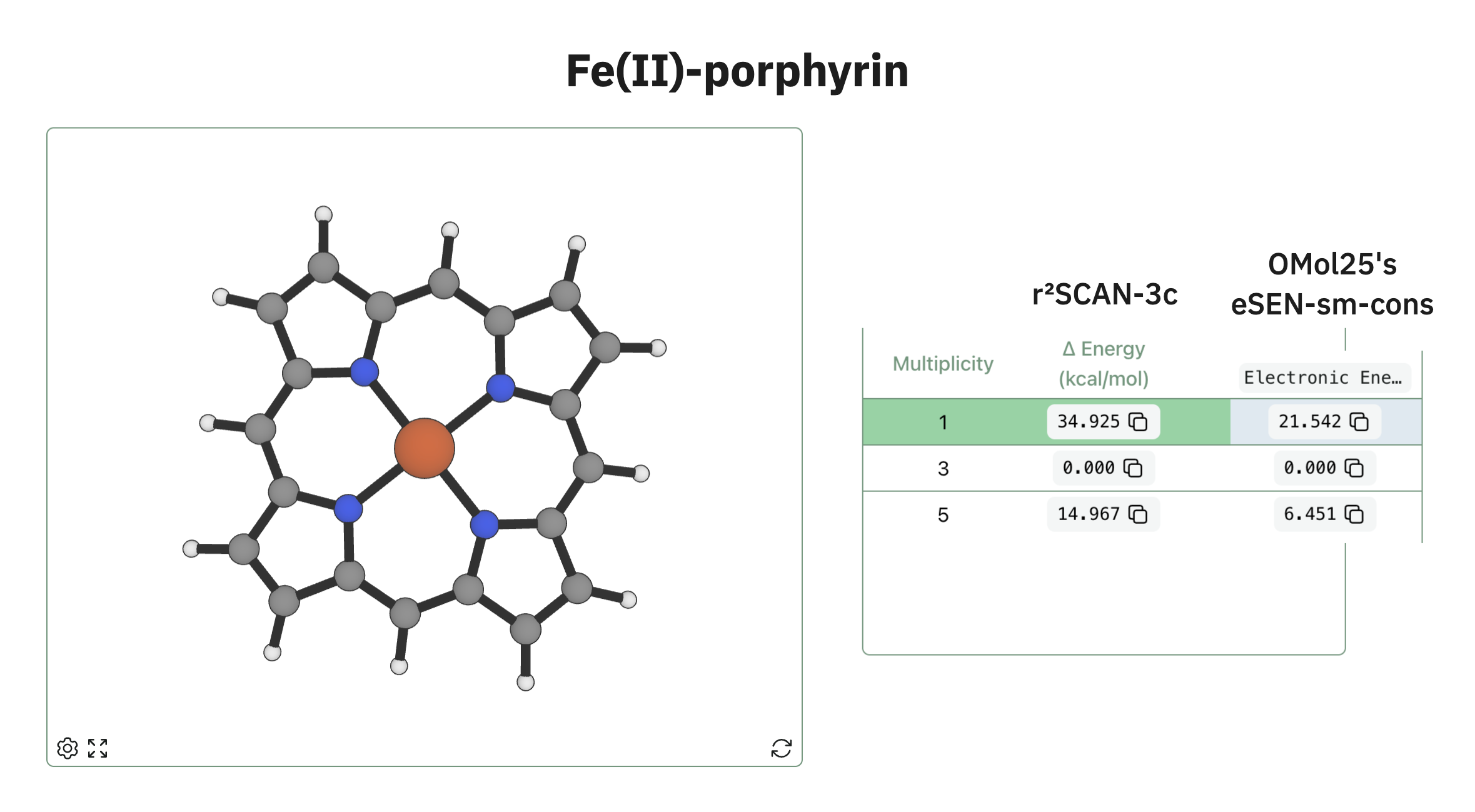
We've already started comparing it to the presets of our spin-states workflow—here's how OMol25's eSEN conserving small model stacks up against r2SCAN-3c for a model porphyrin:
Task-Specific NNPs
We also anticipate that subsets of the OMol25 dataset will be useful in building focused, application-specific NNPs. Large protein fragments like those included in OMol25 have previously been shown to be crucial in building high-accuracy NNPs for studying protein dynamics, while the wide variety of reactive systems included in the dataset might be used to generate reactivity-focused models. While models trained on the whole dataset seem to perform very well, it's likely that smaller models can be considerably more efficient for tasks in which inference speed is crucial, like molecular dynamics on entire biomolecular complexes. (And the dataset is big enough that even 10% of it will be more than enough to build a very good model.)
Implicit Solvent
The models discussed here don't account for solvent effects at all, which limits their applicability to many real-world problems. There are a variety of ways in which one could imagine incorporating solvent effects:
- Run calculations in explicit solvent. This is the most rigorous way to incorporate solvent effects, but also the most challenging. Locating transition states in explicit solvent requires free-energy calculations and enhanced-sampling methods, which quickly makes even the simplest reactions a pain to study. I (Corin) once performed an explicit-solvent study of a relatively simple 2+2 cycloaddition, which ended up taking nanoseconds of MD and a heroic effort across multiple institutions to complete. Improved tooling can doubtless reduce the requisite engineering effort, but this isn't a practical solution for most applications today.
- Incorporate an external physics-based implicit-solvent model. This is what we do here at Rowan for e.g. microscopic pKa calculations; we employ a NNP for gas-phase electronic energy and a separate semiempirical implicit solvent method (often CPCM-X) for solvent corrections. We've had good success with this approach, but it's considerably slower and clunkier than just running a NNP. The implicit-solvent method requires a semiempirical electronic-structure calculation, which can be slow or have convergence issues, and shortcomings in the electronic-structure method are translated to shortcomings in the implicit-solvent energies. (Plus, implicit-solvent methods are notoriously bad for ionic systems.)
- Fine-tune the model on implicit-solvent DFT calculations. This has previously been demonstrated using AIMNet models—retraining the model on DFT calculations run in implicit solvent could produce accurate "solvent-aware" predictions with a fraction of the original training data. This could work well, but requires re-running a lot of DFT calculations in whatever solvents are desired, which gets expensive quickly. Plus, as noted above, implicit-solvent models aren't very accurate for a lot of applications.
- Use an external ML method for solvent corrections. Graph neural networks can be employed as implicit-solvent models through what's essentially a coarse-graining scheme; the average effect of explicit solvent is learned from molecular-dynamics simulations, and then a neural network is used to reproduce the potential of mean force. There's early data showing that these data-driven approaches can be combined with gas-phase DFT calculations to produce good results; why not do the same thing with a NNP? While this is an exciting direction, more work is needed to develop a practical and reliable solution for the full range of applications needed (metals, reactive systems, &c).
Conclusions
We think that Open Molecules 2025 is a big step in the right direction for the field of atomistic machine learning. We're impressed with the scope and scale of the dataset, and we're grateful to Meta and all the researchers involved for spending so much compute to enable the next wave of model building and applied research in this area.
We expect developments will continue to rapidly unfold in this area, and we're sure we haven't yet grasped the full implications of this new dataset. If you're working in this area or just playing with OMol25 and find something interesting, we'd love to hear from you—you can reach us at contact@rowansci.com.
You can already run calculations, scans, intrinsic reaction coordinates, and the like on Rowan with the conservative-force prediction eSEN model. We're planning to continue testing and experimenting with the new pre-trained UMA and eSEN models, with the goal of helping our users apply them to important problems in chemistry, drug discovery, and materials science, and will continue to share what we learn.
Thanks to Daniel Levine, Sam Blau, Kyle Michel, Zach UIissi, and Larry Zitnick for helpful conversations.










