Intramolecular Hydrogen Bonds for Brain-Penetrant Therapeutics
by Corin Wagen · Aug 6, 2024
The blood–brain barrier (BBB) is a semi-permeable membrane that separates the central nervous system (CNS) from the bloodstream. This serves to protect the CNS from toxins and other environmental harms, but also makes it difficult to deliver therapeutics to the CNS: it has been estimated that over 98% of small-molecule drugs are not BBB-penetrant.
Modifications to Increase BBB Penetrance
Extensive empirical studies have found a number of physicochemical properties which typically predict whether a given molecule will be BBB penetrant or not. A study of top CNS-active drugs found that virtually all of them had a low molecular weight, low total polar surface area, high lipophilicity, and—perhaps most crucially—at most one hydrogen-bond donor. These constraints make it extremely difficult to develop potent CNS therapeutics, as polar groups and hydrogen-bond donors are often key design elements that lead to high binding affinity and low IC50 values.
To help make potent drugs more CNS-penetrant without unduly sacrificing potency, hydrogen-bond donors can be "masked" by adding a weak internal hydrogen-bond acceptor to form an intramolecular hydrogen bond. This often increases the permeability of the molecule while maintaining binding affinity: the intramolecular hydrogen bond is robust enough to allow molecules to diffuse through the BBB, but not so strong that the molecule cannot adopt the preferred binding pose and engage a hydrogen-bond acceptor on the target.
Identifying a BBB-Penetrant IP6K1 Inhibitor
A recent paper from Barrow and co-workers at Johns Hopkins used this strategy to design an IP6K1 inhibitor capable of passing through the blood–brain barrier. IP6K1 is a kinase that's key to the inositol phosphate signaling pathway and has attracted considerable interest as a target for many different indications. Sadly, existing IP6K1 inhibitors all contain key carboxylic acids, which are ionized at physiological pH and thus prevent BBB penetration.
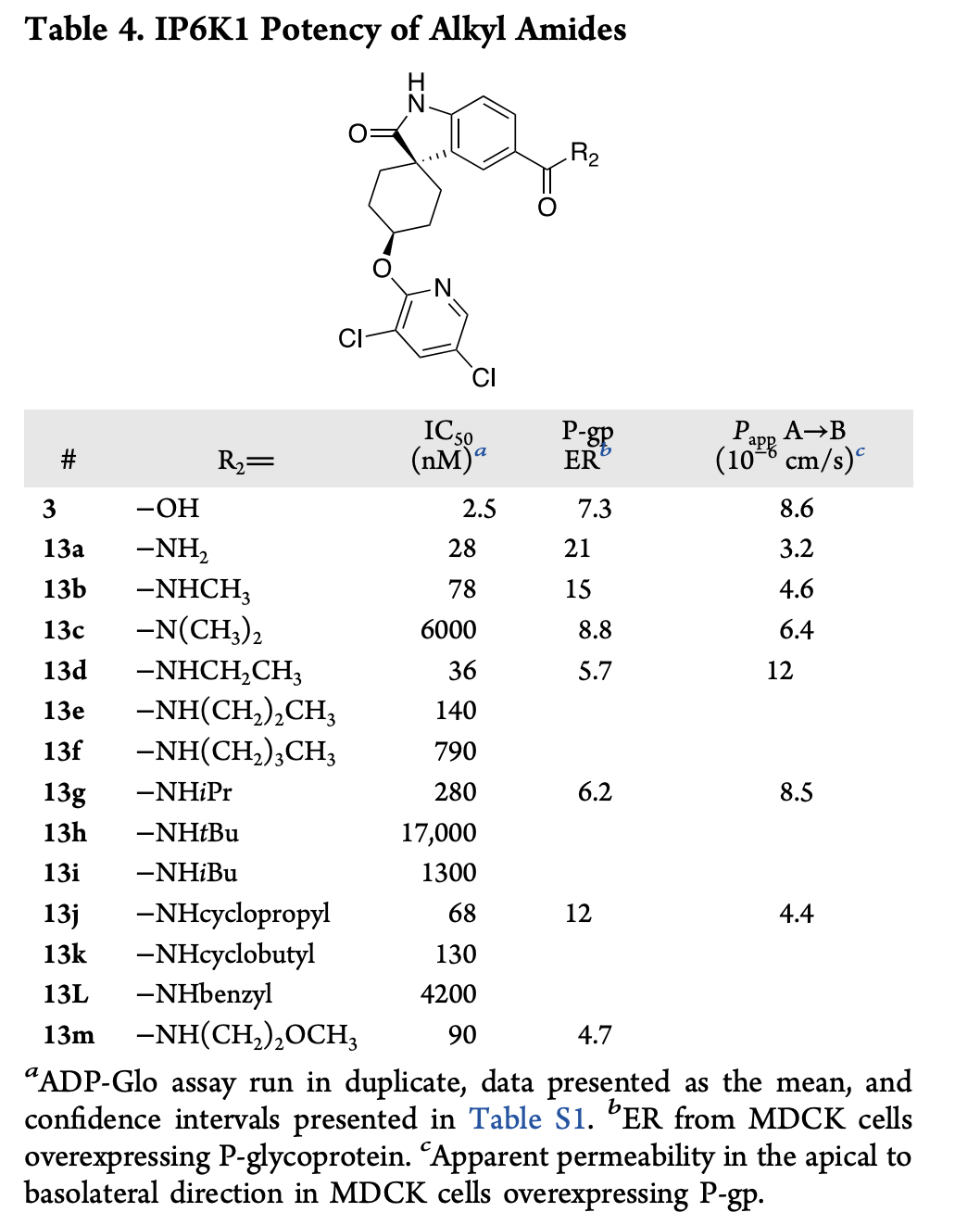
Replacing the carboxylic acid with an amide doesn't improve BBB penetrance
The authors tried a ton of different modifications to turn their existing IP6K1 inhibitor into a CNS-compatible IP6K1 inhibitor. Replacing the carboxylic acid with a carboxamide residue (which is neutral at pH 7.4) preserved most of the potency but led to even lower BBB penetrance, which the authors attribute to the additional hydrogen-bond donor moiety introduced.
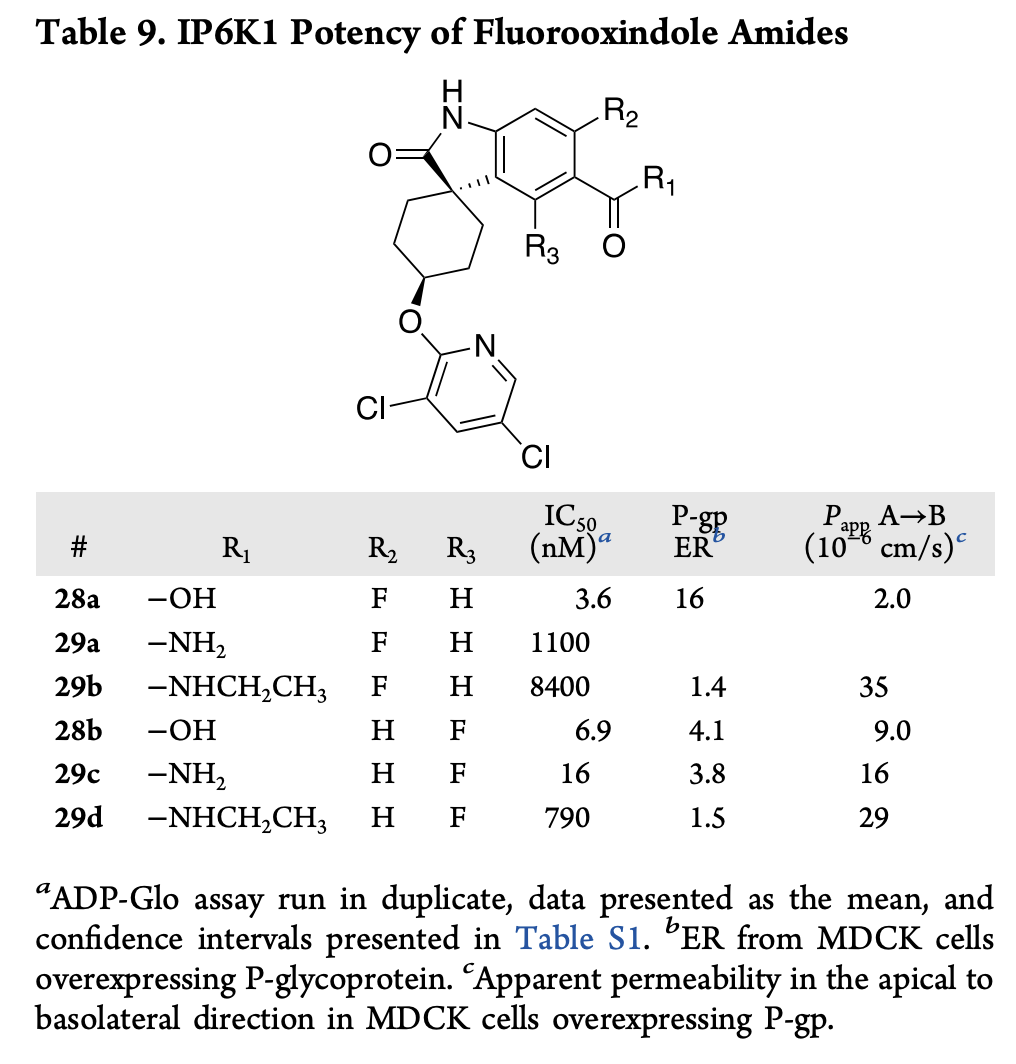
Adding ortho-fluorine atoms dramatically improves permeability
The authors went on to explore introduction of an internal hydrogen bond to mask the carboxamide: while introducing a pyridine group improved permeability at the expense of potency, adding an ortho-fluorine preserved potency and dramatically increased BBB permeability for both the acid and the amide. Ultimately, they chose to move forward with the amide and found that it had a 173x greater brain/plasma ratio than the starting compound and good pharmacokinetic properties.
Modeling Intramolecular Hydrogen Bonding
This paper serves as a great case study of how introducing intramolecular hydrogen bonding can improve BBB penetrance. To understand the effects described in this paper in more detail, we can use Rowan to quickly compute the potential energy surface for the hydrogen-bond acceptor. Here's a comparison of scans for the parent phenyl compound, the pyridyl compound, and the ortho-fluoro compound.
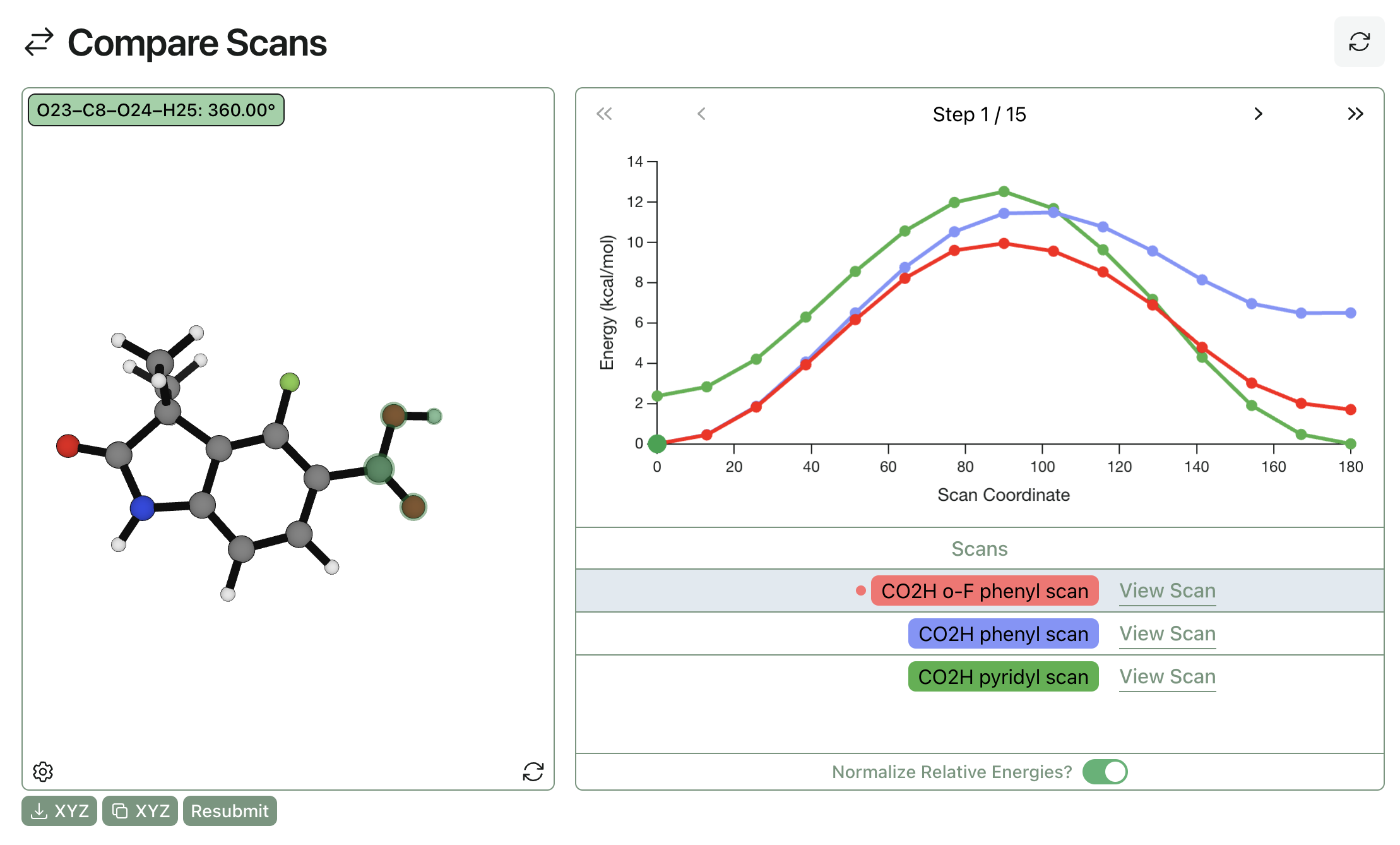
We can see that in the absence of a hydrogen-bond acceptor the s-cis carboxylic acid conformation is strongly preferred. Addition of a strong hydrogen-bond acceptor (pyridine) shifts the ground state towards the s-trans carboxylic acid conformation, consistent with the increased BBB penetrance and decreased potency observed. The ortho-fluorine serves as a weak hydrogen-bond acceptor, making the intramolecular hydrogen-bonded conformation energetically accessible but maintaining the same ground state.
These calculations suggest that there's a "Goldilocks" zone of hydrogen-bond-acceptor strength for intramolecular bonds: acceptors need to be strong enough to mask hydrogen-bond donors in a membrane environment, but not so strong that they compete in the binding site. This chameleonicity has been remarked upon as a key feature of macrocycles, enabling them to change their physicochemical properties in an environment-specific manner.
Prospective Design of BBB-Penetrant Therapeutics
This study, while retrospective, illustrates how calculations can be used for the rational design of CNS-compatible small molecules. The calculations run here took only minutes through Rowan's cloud platform, and the figures were generated automatically through Rowan's molecule viewer.
Rowan provides an optimized workflow for running scans that's ideally suited to running calculations like the ones shown here, where a quick estimate of the relative energies of two states is needed. Rowan also contains cutting-edge machine-learned interatomic potentials that can generate torsional profiles with chemical accuracy in a fraction of the time demanded by traditional quantum chemical methods, meaning that your scans can run in minutes instead of days.










