Property prediction at the boundary of physics and ML
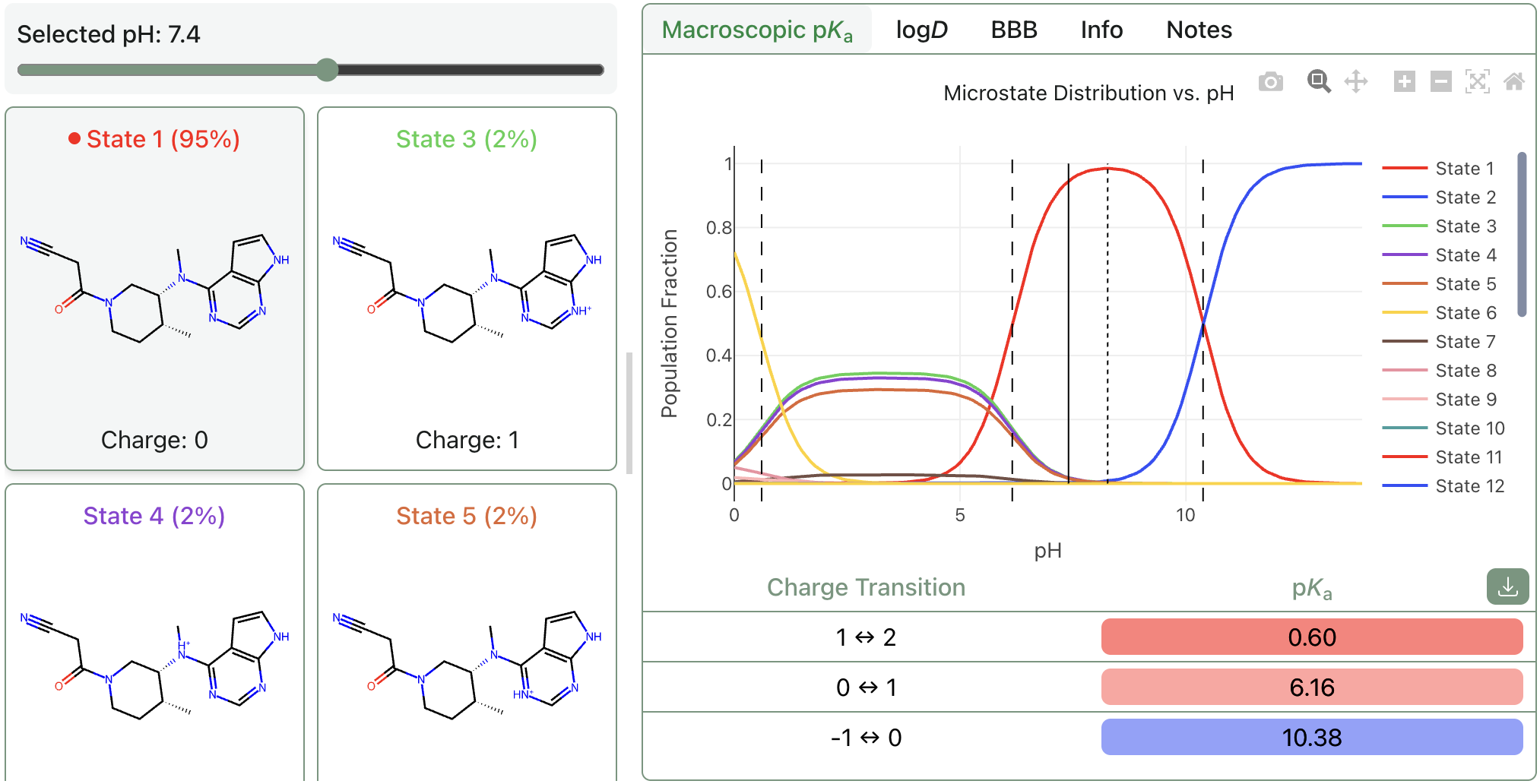

Macroscopic pKa
Predict macroscopic pKa values, microstate populations, isoelectric points, and logD values with Rowan's macroscopic pKa workflow. This is made possible by Starling, a physics-informed machine learning model that runs in minutes. Read the preprint.
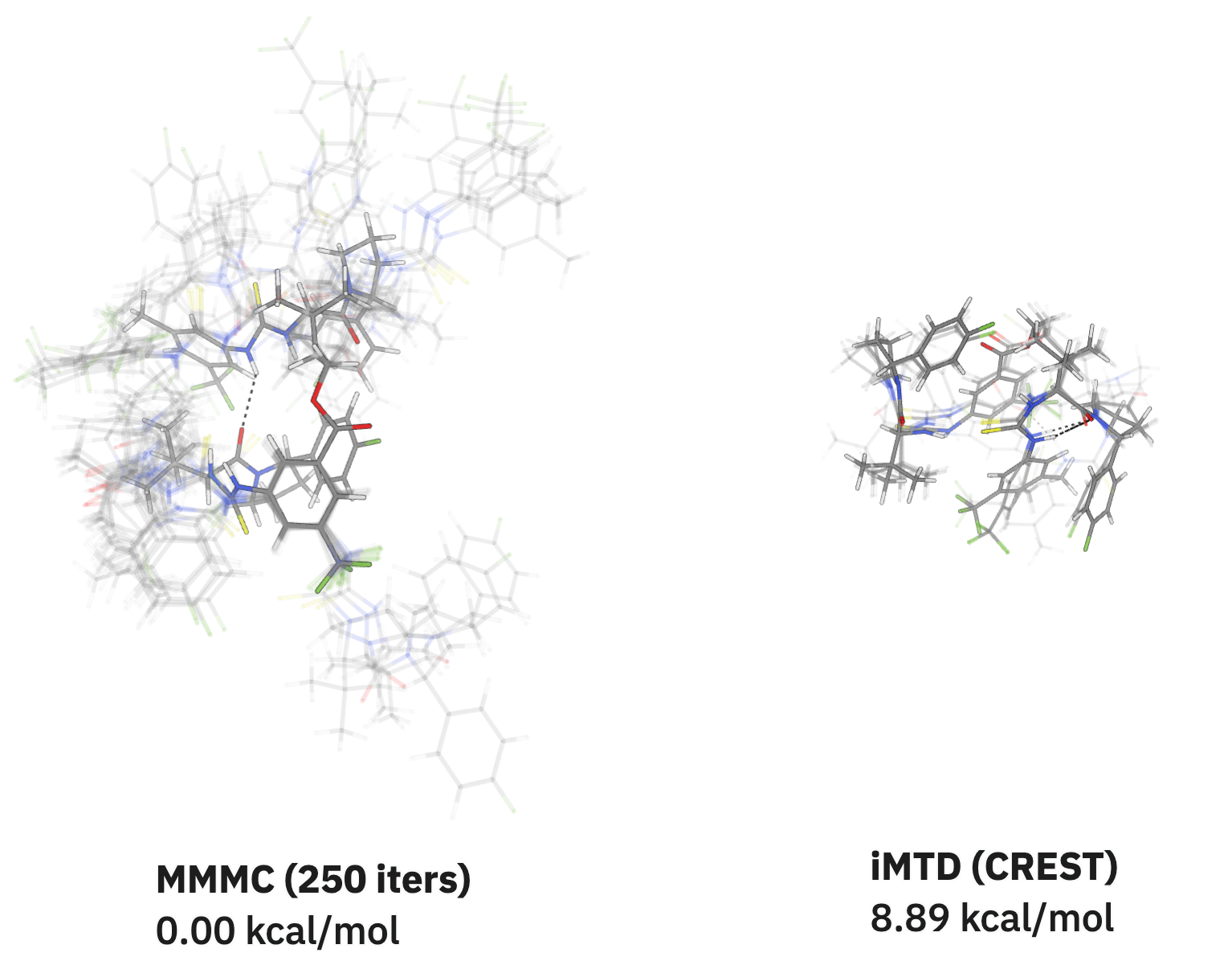
Quick conformer searching
Quickly explore conformational space using fast low-level methods for conformer generation and more accurate final methods for conformer ranking.
Regioselectivity and reactivity
Predict where a molecule will react with nucleophiles, electrophiles, and radicals and quantify an electrophile's ability to promote covalent reactions with Fukui index and global electrophilicity index calculations.
Blood–brain-barrier permeability
Predict the likelihood of blood–brain-barrier penetrance in silico by computing the free energy of neutralization and energy of solvation.
More property predictions
Predict bond-dissociation energies (BDE), solid organic solubility, hydrogen-bond-acceptor strength, redox potentials, and more. Read more about our platform.